二氧化铱是目前商业上唯一可用的质子交换膜电解水制氢的析氧催化剂,对利用可再生能源制备绿氢至关重要。尽管这种催化剂稀有且昂贵,目前尚未找到更便宜的替代品。这是因为大多数催化剂在极酸性条件下会被腐蚀溶解,无法长时间催化。
从原子层面理解二氧化铱催化剂的工作机制,及其为何具有高活性和强稳定性,一方面可以帮助工业界更好地选择铱基催化剂,另一方面也可能为设计更便宜的催化剂提供重要参考。
研究者通常通过四电子转移模型和多种反应中间体来理解析氧催化剂的机理。许多文章参考了丹麦学者Jan Rossmeisl 和 Jen Norskov等人在二十年前利用第一性原理 (DFT) 计算逐步建立的火山图理论模型,来解释不同催化剂的活性。尽管该模型成功地描述了实验结果趋势,但理论计算中所需的众多假设仍难以完全匹配更复杂的实验结果。
近日,帝国理工学院团队利用时间分辨可见光吸收光谱,重新研究了两种常见的二氧化铱催化剂,量化了反应吸附物种,实验验证了理论计算模型,并发现了新的速率控制因素,提出了基于实验的改进的火山图模型。相关成果发表在《Nature Catalysis》和《JACS》上。值得一提的是,最早基于DFT理论计算提出析氧火山图模型的丹麦哥本哈根大学的Jan Rossmeisl教授也是合作和通讯作者之一。
这两篇文章的第一作者都是帝国理工博士生梁才武,Ifan Stephens, James Durrant 和 Reshma Rao为通讯作者。
Nature Catalysis要点精简: 对结晶和非晶氧化铱的结构进行了表征。利用X射线吸收谱发现,结晶和非晶二氧化铱具有相同的[IrO6]八面体结构,只是在结晶结构中这些八面体是长程有序排列,而在非晶结构中是短程有序。利用同位素标记结合时间飞行离子质谱发现,在析氧催化循环过程中,非晶结构中的质子可以渗透到催化剂内部,而结晶结构只在表面进行。这意味着非晶结构中会有远多于结晶结构的Ir电化学活性位点。


通过对光谱的差分分析及线性叠加算法拟合,该工作成功量化了各个氧化还原过程随电压变化的情况。利用DFT计算,研究了 *H2O -> *OH 和 OH -> O 表面物种在结晶结构和类似非晶结构中的转变热力学电压,发现与光谱实验测得的氧化还原电压区间相符。对氧化还原峰进行表面电化学吸附曲线模型拟合,发现该过程更符合Frumkin吸附模型,表明吸附的O物种相互排斥,减弱了O物种的吸附,促进了O-O键的形成。这种相互排斥作用影响了整个反应的动力学。通过吸附模型的拟合,可以得到各个物种在表面的吸附能和相互排斥能,这些势能共同影响着催化物种的反应活性。



最后,利用实验测得的*O的吸附能和相互作用能,作者提出了一种新的火山图理论模型。该模型不仅考虑*O的和催化剂的表面结合能对析氧热力学过电位的影响,还考虑了*O物种之间的相互作用能的影响。根据相互作用能大小变化得到的火山图曲线也会有所不同。在排斥能为0的情况下,该模型与20年前DFT提出的火山图模型一致。研究发现,这种新的模型可以更好地解释实验观测到的不同结构的Ir基催化剂的析氧本征活性。
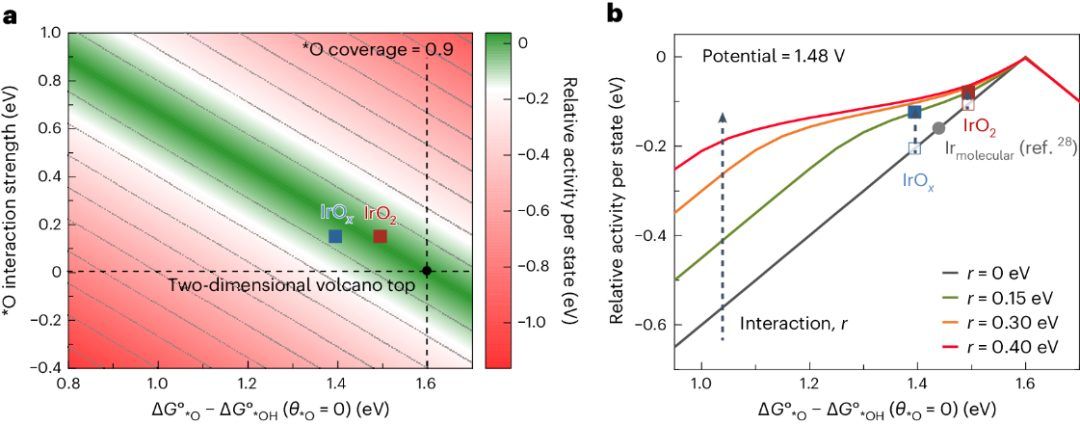
Nature catalysis总结:
该工作结合了实验和理论计算技术,重新评估了水氧化催化剂的设计原理,揭示了活性位点的密度、排列和相互作用对反应速率的影响。发现吸附物种之间的相互作用对水氧化催化速率有显著影响,并基于此提供了比传统DFT表面结合能计算模型更准确的水氧化动力学描述模型,可能为发现更活跃的催化剂提供新的探索途径。
JACS要点总结: 这项工作研究了电解液对电化学界面结构的影响,从而引起的析氧反应动力学的变化。
基于前期的光谱研究,作者结合原位同步辐射X-射线吸收谱,精确确认了Ir在各个氧化还原过程中的化学氧化态转变,发现这些化学价态转变与光谱检测到的变化一致。 在研究中发现,二氧化铱在碱性条件下比在酸性条件下更容易被氧化。通过表面增强原位红外光谱,作者进一步探究了这种现象的原因,发现催化剂-电解液界面的水分子结构不同是关键。在碱性条件下,阳离子与水分子的相互作用使得界面水分子更多呈现不对称结构,这种相互作用使表面氧物种更稳定地吸附在催化剂表面,因此二氧化铱更容易继续氧化并积累更多的氧物种。
作者提出的新火山图模型很好地解释了酸碱条件下二氧化铱的析氧动力学。与传统的DFT计算模型不同,这一模型能够有效评估溶液和界面的影响。

图1:酸性和碱性条件下IrOx的电化学行为

图2:酸性和碱性条件下IrOx的氧化还原特性和浓度随电位的变化

图3:Ir中心的电子结构和配位环境

图4:IrO-电解液界面的水分子结构


图6:活性化学物种的能量学及其对本征水氧化动力学的影响
【展望】
可以想象,这两个工作中提出的新的电化学光谱分析方法不仅适用于二氧化铱催化剂,还可能作为一种通用方法,应用于更多的电催化剂分析,甚至其他电化学系统中的氧化还原反应。只要材料的氧化还原过程能够被可见光光谱捕捉到,这种方法就能发挥作用。
利用这种方法重新理解许多典型的催化剂和反应将会非常有趣,可能会在分子层面上获得新的理解。这种深入的分析将有助于揭示催化剂活性和稳定性背后的机制,进一步推动能源转化催化剂的设计和优化的进程。
Liang, C., Rao, R.R., Svane, K.L. et al. Unravelling the effects of active site density and energetics on the water oxidation activity of iridium oxides. Nat Catal, 2024. https://doi.org/10.1038/s41929-024-01168-7 Caiwu Liang, Yu Katayama, Yemin Tao, Asuka Morinaga, Benjamin Moss, Verónica Celorrio, Mary Ryan, Ifan E. L. Stephens*, James R. Durrant*, and Reshma R. Rao*, Role of Electrolyte pH on Water Oxidation for Iridium Oxides, J. Am. Chem. Soc. 2024. https://doi.org/10.1021/jacs.3c12011
作者信息
第一作者:

梁才武:帝国理工博士生,合作导师Ifan Stephens 教授和James Durrant 英国皇家科学院院士。2017年本科毕业于中南大学材料院,2020年获清华大学材料院硕士学位(导师:杨诚教授)。2021年2月开始在帝国理工攻读博士,主要研究方向为原位时间分辨光谱电化学,水氧化机理解析和PEM电解水制氢研究,截至目前共发表SCI论文17篇, 其中以第一作者(含共一)在Nature Catalysis, JACS, Energy& Environmental Science, Small 等国际期刊发表论文6篇,引用>1700次, H指数13。其中一作文章获评2020年中国百篇最具影响国际学术论文。曾获湖南省优秀毕业生(2017),中南大学优秀毕业生(2017),清华大学优秀毕业生(2020),清华大学优秀硕士毕业论文(2020)等。
通讯作者:

James Durrant, 帝国理工化学系教授,光电物理和光化学家,英国皇家科学院院士,CBE勋章获得者,帝国理工学院可加工电子中心的主任,并且是英国太阳能燃料协会创始人。Durrant 教授从事光,电化学路径下可持续燃料和化学品转化的光谱研究,利用瞬态光谱进行电子和能量转移反应的光化学和光谱电化学表征,从飞秒到秒级时间尺度研究载流子动力学,分析解释材料光电化学特性底层作用机理,并为催化剂和期间提供设计指导。Durrant教授在Nature Energy, Nature Chemistry等期刊发表论文>600 篇,被引超8万次,H-index 156。

Reshma Rao, 帝国理工Grantham 气候变化中心讲师,皇家工程院Fellow, 博士毕业于麻省理工学院,师从Shao-Horn Yang教授。博后于帝国理工大学,师从James Durrant教授。曾获Clara Immerwahr 奖, 英国亚洲女性成就奖。Reshma主要研究聚焦于利用原位光谱技术研究固-气和固-液界面的(电)化学反应,理解反应机理,包括原位同步辐射X-射线谱,表面增强红外光谱,可见光谱等。通过对催化机理的理解,她致力于开发高活性、高稳且低成本的能源转化催化剂。目前在Nature Catalysis, JACS, Joule 等期刊发表论文>50篇,被引>5800次,H-index 27。

Jan Rossmeisl 丹麦哥本哈根大学化学系教授,高熵合金催化中心主任,Jan Rossmeisl在理论催化领域做出重要贡献,其中他参与建立的DFT计算模型是目前广泛应用于理论电催化领域快速估算电极材料过电位的有效方法。Rossmeisl教授主要研究领域:理论电催化,非均相催化,光催化,原子尺度模型,密度泛函理论,反应动力学,能量储存和转换。目前在Science, Nature chemistry, Nature material, Energy & Environmental Science等期刊发表论文>200篇,被引 >7万次, H-index 104。

Ifan Stephens, 帝国理工学院材料系电化学教授, 电化学家,曾任丹麦技术大学副教授,麻省理工访问副教授,获RSC John Jeyes 奖,HP Now公司创始人之一。Stephens教授主要从事可再生能源的大规模电化学转换过程中电化学界面的理解和利用,这包括氮还原制氨,氧还原,电解水,二氧化碳还原,和电池气态产物原位检测等。Ifan 的研究发现了多种新的高效的氧还原催化剂,目前在Science, Nature 等期刊发表论文>150篇,被引2.3万+,h-index 56。
