大环肽配体的系统优化是一个严峻的挑战。在这里,我们描述了一种使用细胞蛋白系统进行先导化合物优化的方法,作为从初始先导化合物转向临床候选药物的回顾性示例。我们展示了如何通过利用 核磁 数据来识别先导化合物的低能溶液整体来导出构象限制。

这种限制可用于集中对类似物的构象搜索,以便通过分子对接准确预测结合的配体姿势,从而估计配体应变和蛋白质-配体分子间结合能。我们还描述了一种类似的基于配体的方法,该方法采用分子相似性优化来预测结合姿势。
 大环构象的重要作用
大环构象的重要作用这两种方法都被证明对于优先考虑先导化合物类似物是有效的。相对较小的配体修饰可能对预测的结合位姿或分子间相互作用影响最小,但通常会导致估计应变的巨大变化,从而对总体结合能估计产生主导影响。

有效的大环构象搜索至关重要,无论是在基于 核磁 的约束、X 射线配体细化、用于对接/配体相似性计算的部分扭转约束还是对标称全局最小值的不可知搜索中。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法。

可以使肽大环化合物的先导化合物优化更加高效。通常会导致估计应变的巨大变化,从而对总体结合能估计产生主导影响。有效的大环构象搜索至关重要,无论是在基于 核磁 的约束、X 射线配体细化、用于对接配体相似性计算的部分扭转约束。

还是对标称全局最小值的不可知搜索中。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法,可以使肽大环化合物的先导化合物优化更加高效。通常会导致估计应变的巨大变化,从而对总体结合能估计产生主导影响。

有效的大环构象搜索至关重要,无论是在基于核磁的约束、X 射线配体细化、用于对接/配体相似性计算的部分扭转约束还是对标称全局最小值的不可知搜索中。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法。

可以使肽大环化合物的先导化合物优化更加高效。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法,可以使肽大环化合物的先导化合物优化更加高效。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法,可以使肽大环化合物的先导化合物优化更加高效。
 优化先导化合物
优化先导化合物使用现代 展示技术对体外表达的大环肽进行基于亲和力的选择可以识别相对有效和选择性的先导化合物。然而,大环肽配体的系统优化是一个严峻的挑战。在这里,我们描述了一种使用 细胞蛋白系统优化此类先导化合物的方法。

作为从初始先导化合物转向临床候选药物的回顾性示例。我们展示了如何通过利用 核磁 数据来识别先导化合物的低能溶液整体来导出构象限制。2016 年公开的一份专利申请中公开了一种 细胞蛋白先导化合物和众多类似物。

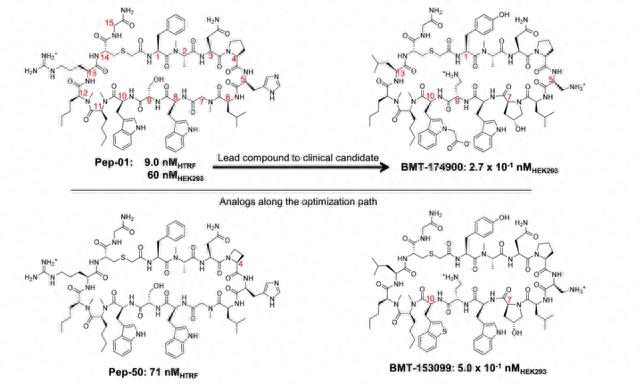
在从最初的先导化合物转向临床候选化合物的过程中,需要对大环肽中的6个位置进行修改。这是通过基于结构的药物设计实现的,需要合成和评估数千种化合物的迭代过程。该过程由大环配体与 细胞蛋白 的多个共晶结构引导。

但 BMT-174900 的路径并未广泛使用较小非大环分子常用的计算方法类型。图1中的示例结构 对微小的结构变化表现出高度的敏感性。正如我们将看到的,这些活性的巨大变化只能部分地通过蛋白质-配体结合相互作用来解释,其中构象能量学的变化起着至关重要的作用。

我们分析了最近开发的大环配体建模方法在未来此类先导化合物优化项目中的应用程度。在过去的几年中,大环配体的计算建模方法取得了重大进展。使用将生物物理数据与实用且高效的计算方法相结合的多学科方法,可以使肽大环化合物的先导化合物优化更加高效。

特别是,基于天然产物和半合成的大环化合物,其总可旋转键多达大约 21-23 个,在利用多个构象搜索的准确性和速度方面已被证明是易于处理的计算核心。然而,更大的肽大环仍然具有挑战性,特别是在跨环氢键的“阶梯”不形成稳定网络的情况下。

为了比较,所示的例子 每个都有 60 个或更多的总可旋转键——远远超出了没有生物物理数据来减少搜索空间的可处理范围。最近,我们展示了如何使用从 核磁 测量得出的距离和二面体约束来阐明肽大环的低能量解系综。

图 2说明了如何从 核磁 限制的构象搜索或从 X 射线晶体学以及仔细细化结合的大环坐标衍生出优选的大环构象。在许多情况下,获得足够质量的 X 射线共晶结构可能是难以克服的。对于精心选择的大环结构,溶液状态通常反映了很大程度上朝向束缚态的预组织。
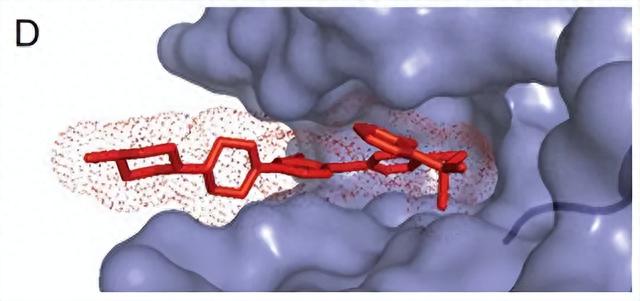
从拟合良好的构象到 X 射线密度或来自满足 核磁 限制的低能构象异构体池的代表性范例,子结构可用于定义构象偏好。底部的子结构是从以洋红色显示的 核磁 溶液系综的最低能量构象异构体中提取的。

图 3说明了如何使用构象偏好来指导构象搜索以预测新类似物的结合态。可以通过将提议的类似物与分子片段进行图形匹配来实现对这种偏好的遵守。新类似物和给定片段之间的子图匹配用于实例化扭转约束以匹配片段的构象。

通过使用方井二次能量惩罚来施加约束,该能量惩罚允许在对与优选扭转角的偏差的一定容差内零惩罚。结构生成是在给定的限制下完成的,构象搜索是在有或没有限制的情况下完成的。对于与扭转约束相匹配的分子部分,发生相对较小的构象变化。

对于分子中不匹配的部分,根据能量学的考虑,可能存在很大的变化。受限构象异构体系综用作分子对接或分子相似性计算的输入,以预测类似物的结合姿势。 对接或基于相似性的优化过程中发生的位姿优化会产生集中的绑定构象异构体集合。

无约束系综用于识别全局最小能量。利用大环构象偏好来预测结合姿势的方案,可以使用对接或配体相似性。对于基于配体的评分,将常数值添加到估计的应变能中,以便将两个协议的评分放在相同的粗略范围内。

从而得出蛋白质-配体结合能的焓分量的最终估计。基于结构的方案受益于为精心设计的类似物识别与蛋白质的新相互作用的能力。对于纯粹基于配体的方案,将类似物的受限构象异构体整体与基于 核磁 的先导化合物溶液整体的示例对齐。

这种基于相似性的对齐用于结合姿态预测,提供与基于结构的协议中获得的类似的结合构象能量值。请注意,当寻求效力显着增加时,标称相似性得分值可能无法用于化合物排名,这需要偏离先导化合物。

在寻求保持效力同时使基础化学结构多样化的设计方案中,相似性分数值可能有用。但在这项工作中,仅使用相似性优化过程产生的姿势,而不是使用相似性得分。令人惊讶的是,相对较小的配体修饰可能对预测的结合姿势或分子间相互作用影响最小。

但通常会导致估计应变的巨大变化,从而对总体结合能估计产生主导影响。在这项工作中,估计配体应变的变化解释了测量活性变化的最大部分。束缚态和解态的能量估计差异的重要性使得有效的大环构象搜索显得尤为重要。

无论是在基于 核磁 的约束、X 射线配体细化、用于对接或配体相似性计算的部分扭转约束还是对标称全局最小值的不可知搜索的背景下,构象搜索都必须彻底且高效。接下来,对大量初始先导化合物的类似物进行刚刚描述的。

基于结构和基于配体的工作流程的回顾性应用。虽然利用蛋白质结构的计算提供了更多信息,但由于从结合配体应变的估计中看到的巨大影响,纯粹基于配体的工作流程可能很有价值。
 笔者认为
笔者认为将描述应用两个计算工作流程来优先考虑先导化合物 类似物的结果:一种基于结构的方法,需要顺应构象的 细胞蛋白 晶体结构来结合该系列中的大环化合物,以及纯粹基于配体的方法。两种方法都利用信息来部分约束需要搜索的构象空间以预测绑定姿势。

使用深度 方法进行了非常彻底的构象搜索。有关 核磁 实验方面和构象搜索方法的更多详细信息,请参阅方法和数据。图显示了 细胞蛋白 结合态与来自核磁限制构象搜索程序的整体中最匹配的构象异构体之间的比较。
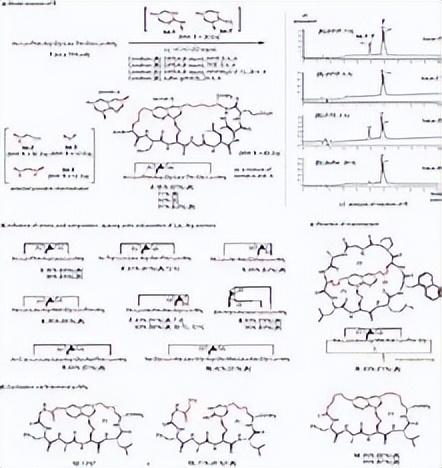
单个最低能量 构象异构体,标记有氢键来自最低能量构象异构体的五个替代分子亚片段。显然,溶液状态是为了结合 细胞蛋白 而预先组织的。特别是,埋藏侧链在解决方案整体中显示出相对较小的移动。
 参考文献
参考文献《用于发现伪天然大环肽》化学研究学会
《隐式低模式速度过滤应用于大环和蛋白质环的构象搜索》化学信息模型
《进行大环构象采样》 化学信息模型
