华算科技结构优化结构优化不收敛至少包含两类问题:电子自洽不收敛和离子结构不收敛。前者表现为 SCF 能量振荡、磁矩跳变或电荷密度难以稳定;后者表现为能量下降很慢、力始终较大、原子来回摆动或晶胞畸变异常。混淆两类问题会导致错误处理。
式中 Fi 是第 i 个原子受到的力,E 是体系总能,Ri 是原子坐标。常见收敛标准为最大力低于 0.01–0.05 eV Å−1,高精度声子或弹性计算通常需要更严格阈值。

导入时还应检查对称性和占位。实验 CIF 中可能含有部分占位、无序溶剂或平均结构,直接转成 DFT 模型会产生非整数原子数或异常短接触。对这类结构,先构建有序超胞并合理分配占位,比直接按原始 CIF 优化更稳妥。
对于表面和界面模型,真空层不足会造成周期镜像相互作用,通常需要至少 15–20 Å 真空;二维材料若同时放开 c 轴晶胞,程序可能把真空当成可压缩空间处理。分子吸附体系还应避免偶极矩方向上的虚假电场,必要时加入偶极修正。
约束设置也会制造假性不收敛。固定层太少会让表面整体漂移,固定层太多又会锁死真实弛豫;选择性动力学标签写错时,某些原子看似无法降低受力。检查优化轨迹比只看最终报错更可靠。
三、电子步振荡
金属、窄带隙半导体、强关联氧化物和磁性体系容易出现 SCF 振荡。原因是电荷密度更新过猛,导致每一步都在不同电子构型之间跳动。降低混合参数、增加展宽、使用更稳健的初猜电荷,常比单纯增加最大电子步更有效。
吸附优化中 Eads 的可靠性依赖三个结构都充分收敛。若只有复合体系放松,而清洁表面或分子未达到同等标准,吸附能误差可达到 0.1 eV 以上,足以改变催化活性排序。

当优化失败出现在晶胞自由度上,应单独检查压力张量。层状材料、分子晶体和多孔材料对范德华作用很敏感,未加色散校正时层间距可能持续膨胀;压力收敛阈值设得过严,也会让晶胞在微小应力下反复振荡。
当优化持续不收敛但能量已经接近平坦时,需要判断是否存在真实不稳定。可计算声子虚频、检查弹性常数或沿软模方向扰动再优化。若体系本来处在相变路径、扩散过渡态或吸附解离过程中,强行寻找局域极小点可能不符合研究目标。
若研究对象是反应路径,还需要区分极小点优化与过渡态搜索。NEB 初始链中的中间构型常常力较大,不能按普通结构优化标准逐个要求收敛到极小点;真正需要检查的是路径连续性、最高能点附近切向力以及最终过渡态虚频。
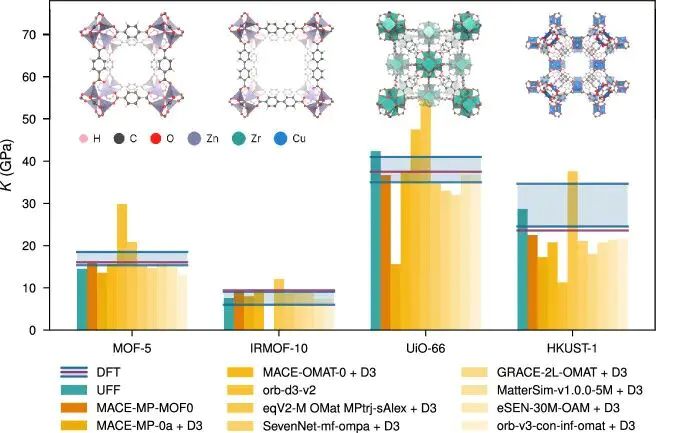
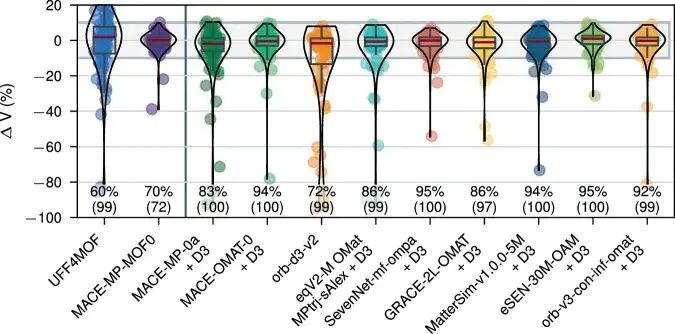
图5:MOF 结构建模与势能评估流程体现结构弛豫、力场近似和优化稳定性的关系。DOI:10.1038/s41524-025-01872-3。