编者按:人类基因组计划开启了生命科学的新纪元,而人工智能(AI)与合成生物学的飞速发展,正在将基因组研究从“读取”推向“注释”与“书写”的全新维度。在2026年病毒性肝炎慢性化重症化基础与临床研究进展学术会议上,重庆医科大学感染性疾病分子生物学教育部重点实验室黄爱龙教授在主题报告中,系统阐述了“乙型肝炎病毒(HBV)万例全基因组深度解码计划”(Hex-10K)的核心理念、技术路径与科学价值,为乙肝功能性治愈研究提供了全新的基因组学视角。
 从人类基因组计划到HBV基因组解码:科学范式的演进
从人类基因组计划到HBV基因组解码:科学范式的演进人类基因组计划是生命科学史上最为宏大的工程,被视为与“阿波罗登月计划”并列的科学壮举。这项历时13年、耗资30亿美元的国际协作工程,动用了全球数百名顶尖研究人员,最终在2001年发布了人类基因组草图,并在2003年宣布完成。人类基因组计划的成功并非偶然,而是人才、科学思想、技术实验方法、资源组织以及管理模式这四个核心要素在时机成熟时的必然交汇。
在该计划取得成功的基础上,千人基因组计划于2007年启动,旨在通过对来自多个不同人群的多样化个体进行全基因组测序,全面描述人类常见的基因变异情况。该项目于2015年公布了来自不同种族背景的超过2504名个体的基因组,使我们对决定生理特征的遗传因素有了大量了解(图1)[1]。然而,遗传如何决定性状,不仅与传统遗传学相关,更与表观遗传学密切相关,涉及复制、转录、翻译、结构与修饰等过程。2003年启动的DNA元件百科全书计划,目标是建立一个详尽的人类基因组功能元件百科全书,包括基因、与基因调控相关的生化区域(如转录因子结合位点、开放染色质和组蛋白标记)和转录异构体,将研究视野从编码区域拓展至非编码区域[2]。
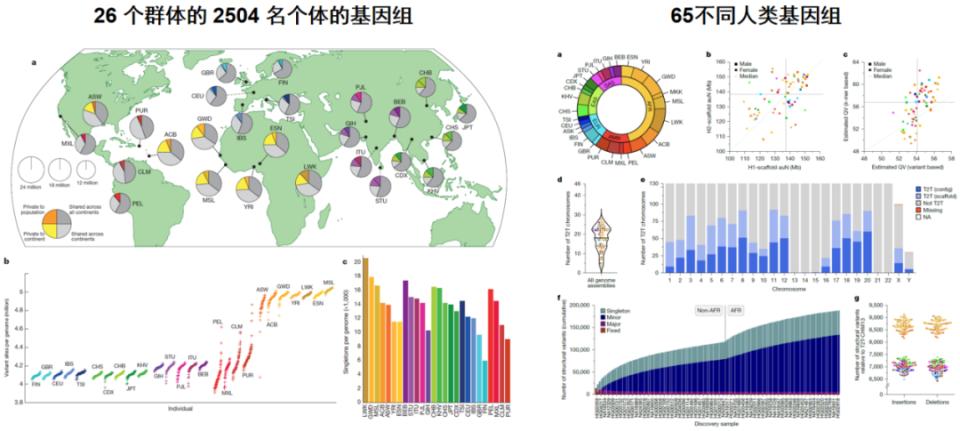
图1. 人种遗传特征
(引自讲者会议幻灯)
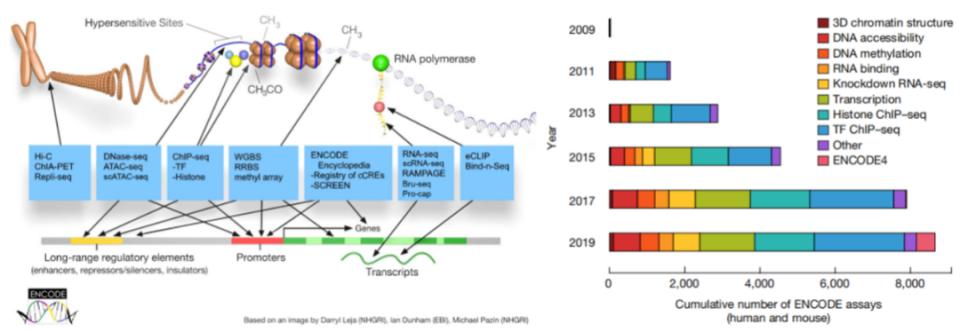
图2. 代码编辑语法——DNA元件百科全书(ENCODE)
(引自讲者会议幻灯)
2012年,英国政府提出了“十万基因组计划”,重点关注罕见病、部分常见癌症以及传染病。该计划对约85000名患有罕见疾病或癌症的患者进行10万人份基因组测序,招募工作于2018年12月完成。通过对13880例实体瘤(涵盖33种癌症类型)患者进行全基因组测序分析,研究人员将突变特征、拷贝数畸变、结构变异、同源重组缺陷和肿瘤突变负荷等数据与患者的5年常规临床数据相整合,发现癌症中特定的基因变化与生存率、患者预后密切相关,证明了将基因组数据和真实世界临床数据相结合的实用性[3]。
人工智能驱动的基因组解码:从代码读取到功能注释海量的基因组解码数据若没有强大的计算工具支撑,将仅是一堆冗余的字符。生物信息学和人工智能的发展使得“生命”能够以数字的形式在硅片上运行。DNA基础模型包括Nucleotide Transformer和Evo等,Nucleotide Transformer通过掩码语言建模学习基因组信息,而Evo利用状态空间模型获得更长的模型上下文长度,以更有效地发现长序列中的模式。RNA基础模型中,RNAErnie使用多阶段掩码策略和基于类型的微调提升对RNA序列的理解能力,而DGRNA等模型则通过更大的数据集和更新的模型架构提升预测性能。蛋白质基础模型包括ProGen系列和ESM系列等,ESM系列在结构预测等任务上表现突出,与AlphaFold等方法相比,ESMfold精度稍低但速度更快[4]。
AlphaGenome模型的诞生标志着基因组学领域的革命性突破。该模型能够处理高达100万碱基对的长DNA序列,并以前所未有的精度和广度预测人类DNA序列中的单碱基突变如何影响调控基因的多种生物过程。AlphaGenome不仅能帮助研究人员更精确地定位疾病的潜在根源,阐释与特定性状相关的变异所造成的功能性影响,从而有望揭示新的治疗靶点,还可用于指导设计具备特定调控功能的合成DNA,推动未来的生物学基础研究[5]。
启动子活性调控模型是一个经济高效的深度学习框架,通过结合定制化大规模并行报告实验数据与轻量级卷积神经网络,实现了仅从DNA序列直接预测人类启动子活性,并系统解析了其在多种细胞类型及刺激响应下的转录因子调控语法。该模型以单核苷酸分辨率学习转录起始位点周围的组蛋白修饰、DNA可及性、转录因子结合及链特异性基因表达等功能基因组学特征,构建启动子序列与调控表型的基础关联,首次实现了对启动子变异表达效应的精准预测[6]。
合成基因组学:从读取到编辑当基因组解码达到极致,接踵而至的便是基因组的书写与合成。合成基因组学标志着人类不仅能读懂生命的指令,还能从头构建具有特定功能的生命系统。2002年,纽约州立大学的科学家成功合成了脊髓灰质炎病毒,展示了“从无到有”智造病毒的潜力。随后,CRISPR-Cas9等技术的发展,为病毒基因工程的研究提供了高准确性和高效率的工具技术[7,8]。
Nature杂志的焦点报道指出,科学家们可利用人工智能编写病毒基因组,然后制造出杀死细菌的病毒。DNA测序和合成技术的进步极大地提升了我们在全基因组尺度上读取和编写DNA的能力。斯坦福大学化学工程系团队开发出的基因组语言模型Evo,通过排除特定数据,能够成功阻止模型设计真核病毒,包括致病性人类病毒[9]。
选择HBV基因组的依据:小而精悍的天然模型从复杂系统到微观战争,当我们掌握了海量的解码工具与计算能力后,我们需要一个“天然的基因组模型”来验证这些技术,并解决人类最迫切的健康危机。相比于人类30亿碱基对的复杂性,病毒基因组小而精悍,是应用“AI for Science”实现从“解析”到“功能模拟”再到“治愈设计”的最佳实验场。
我国慢乙肝防控任务仍然紧迫和繁重。我国仍是乙肝危害最严重的国家,HBV感染者7500万,占全世界的27%,每年乙肝相关疾病死亡16万,占全世界的29%。疫苗接种有效控制了新发感染(5岁以下感染率0.38%),但存量感染是当前的主要矛盾,矛盾的解决只能靠治愈[10,11]。
HBV基因组具有种属特异性、组织特异性和年龄依赖性特征,为不完全闭合双链DNA,约3.2kb,包含4个开放读码框,编码7种病毒蛋白(图3)。HBV的功能性治愈本质在于对cccDNA的功能抑制。cccDNA是子代病毒产生的起始物,是HBV感染持续、难以治愈的根本原因,其半衰期长达数月,而现有药物对cccDNA的影响很有限[12]。
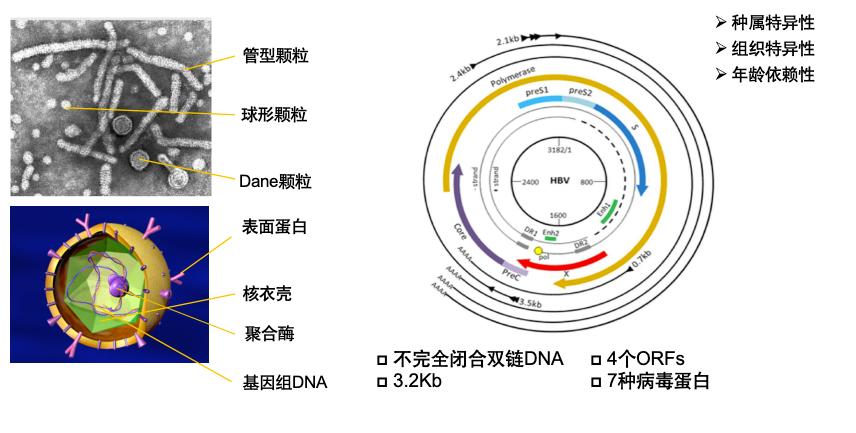
图3. HBV的结构与基因组特征
(引自讲者会议幻灯)
HBV全基因组解码计划(Hex-10K)的构建与初步成果基于上述背景,黄爱龙教授团队正式提出“HBV万例全基因组深度解码计划”(HBV Extensive X-Decoding 10K Project,简称Hex-10K)。项目名称中“Hex”代表十六进制,是计算机科学的底层语言,直接宣示该项目是原生AI驱动的科研项目,暗示深度学习、神经网络算法在全基因组解码中的核心地位;同时六角形在自然科学中是最稳定、最完美的填充结构,象征着项目对海量变异位点的解析具有极高的稳定性和精准度。“Ten”对应10K(10,000)样本量,在科研界样本量上万是质变的分水岭,意味着该项目拥有极高的统计效力和捕捉罕见变异的能力。
项目分三阶段进行:读取、注释、编辑,对应过程包括语料、语义、语法。第一阶段包括数据生产体系、质量控制体系和分析流程,已完成基础设施建设的概念验证(0-1)。团队已建立了规范的临床样本收集及测序流程,构建了HBV基因组测序和序列预处理分析流程,通过Snakemake进行流程化管理。在基因型预测方面,准确率达到100%。团队绘制了HBV基因组突变图谱,识别出多个高频突变位点,如1726位A→C、915位A→G、1838位A→G、2738位A→G、457位C→A、1389位C→T、540位G→A、1896位G→A、1969位T→C等。经初步分析,在HBeAg血清学转换过程中,除外BCP区A1762T/G1764A双突变及PC区G1896A可导致HBeAg分泌减少或合成终止,基因组蕴藏多种e抗原转阴机制,这一发现与临床表型密切相关。此外,团队还分析了年龄和HBV滴度分布特征,为后续研究提供了基础数据。
团队正在构建HBV专属多模态基础模型,但同时也认识到当前研究的局限性:低病毒载量患者尚未覆盖,干预因素(用药情况)可能掌握不全,自然感染队列和宿主因素暂未考虑等。
展望:从解码到治愈HBV基因组虽小,却浓缩了生命演化最复杂的逻辑,也承载着我国数亿家庭的健康负担。Hex-10K计划试图在3.2kb的微观世界里,打赢这场精准医学的持久战。通过整合大规模临床样本、深度测序技术和AI驱动的多模态基础模型,该计划有望系统揭示HBV基因组的完整变异图谱,阐明关键突变与疾病进展、治疗应答的关联,为乙肝功能性治愈的精准靶点发现和个体化治疗策略制定提供坚实的基因组学基础。
参考文献:
1.The 1000 Genomes Project Consortium., Nature. 2015.
2.ENCODE Project Consortium., Nature. 2012.
3.Sosinsky et al., Nature Medicine, 2026.
4.Li et al., Frontiers of Digital Education, 2025.
5.Avsec et al., Nature, 2025.
6.Jaganathan et al., Science, 2025.
7.Fu et al., ACS Synth Biol, 2024.
8.Richard Allen et al., msystems, 2021.
9.Katie Kavanagh, Nature. 2025.
10.Lancet 2023.
11.Lancet Reg Health West Pac 2024.
12.Cohen et al, Science. 2018.