Scissor的安装真的很麻烦,但是数信院共享服务器已经帮你预装好了,还同时支持Seurat v4和Seurat v5的对象~
都2025年啦!我们甚至不用详细统计也能知道scRNA-seq和bulk RNA-seq技术已经在生命科学的各个领域都产生了海量的数据。虽然scRNA能够提供单细胞级别的基因表达信息,但是其基因测序深度又是非常浅的,从而导致下游任务如聚类注释的局限。因此我们该如何结合海量的bulk RNA数据以提高scRNA分析的分辨率和鲁棒性?
高认可度算法1——scAB
发表于NAR[scAB detects multiresolution cell states with clinical significance by integrating single-cell genomics and bulk sequencing data],文章Fig1其实清晰展示了算法的核心思想,输入为scRNA或scATAC数据和bulkRNA数据,然后进行类似[矩阵分解]的操作,输出为W矩阵[行为bulk样本,列为模块]和H矩阵[行为模块,列为单细胞],最后就可以基于H矩阵进行下游推断任务——分群/通路富集/生存分析等。
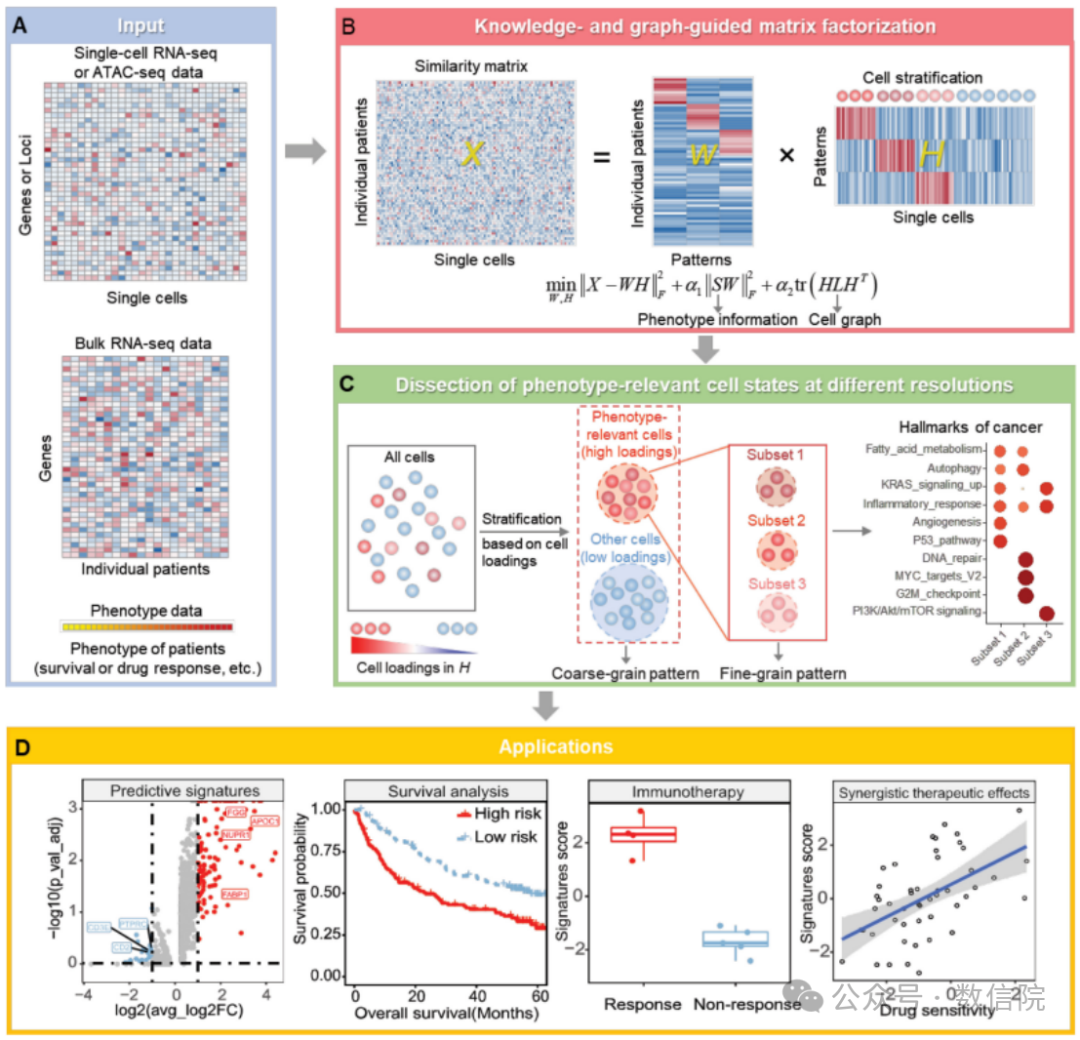
Figure1
scAB的代码实现——数信院共享服务器已经预装,上号即用
安装
devtools::install_github("Qinran-Zhang/scAB")加载包和测试数据library(Seurat)library(preprocessCore)library(scAB)data("data_survival")dim(sc_dataset)#> [1] 21287 8853head(bulk_dataset[,1:10])#> TCGA-2Y-A9GS-01 TCGA-2Y-A9GT-01 TCGA-2Y-A9GU-01 TCGA-2Y-A9GV-01#> ARHGEF10L 11.5199 12.2544 12.6434 11.9114#> HIF3A 2.4425 4.6559 1.8083 3.7751#> RNF17 3.7881 0.0000 0.0000 0.0000#> RNF10 11.5109 11.7863 12.3187 11.7253#> RNF11 10.6574 11.0133 10.5722 10.9586#> RNF13 10.2623 10.4574 9.9284 10.5863#> TCGA-2Y-A9GW-01 TCGA-2Y-A9GX-01 TCGA-2Y-A9GY-01 TCGA-2Y-A9GZ-01#> ARHGEF10L 11.8108 10.6616 10.6321 12.4218#> HIF3A 2.8340 3.2456 7.1429 5.7972#> RNF17 0.0000 0.0000 7.9222 0.7507#> RNF10 11.8382 11.5872 11.5770 11.2699#> RNF11 10.3071 10.7500 9.9313 10.7820#> RNF13 10.1785 10.4880 10.0815 10.5819#> TCGA-2Y-A9H0-01 TCGA-2Y-A9H1-01#> ARHGEF10L 10.8164 10.2715#> HIF3A 3.7561 4.3687#> RNF17 0.0000 0.0000#> RNF10 12.7789 11.2257#> RNF11 10.0653 10.1549#> RNF13 10.6751 9.9593head(phenotype)#> time status#> TCGA-2Y-A9GS-01 724 1#> TCGA-2Y-A9GT-01 1624 1#> TCGA-2Y-A9GU-01 1939 0#> TCGA-2Y-A9GV-01 2532 1#> TCGA-2Y-A9GW-01 1271 1#> TCGA-2Y-A9GX-01 2442 0
单细胞数据分析
sc_dataset <- run_seurat(sc_dataset,verbose = FALSE)UMAP_celltype <- DimPlot(sc_dataset, reduction ="umap",group.by="celltype")UMAP_celltype
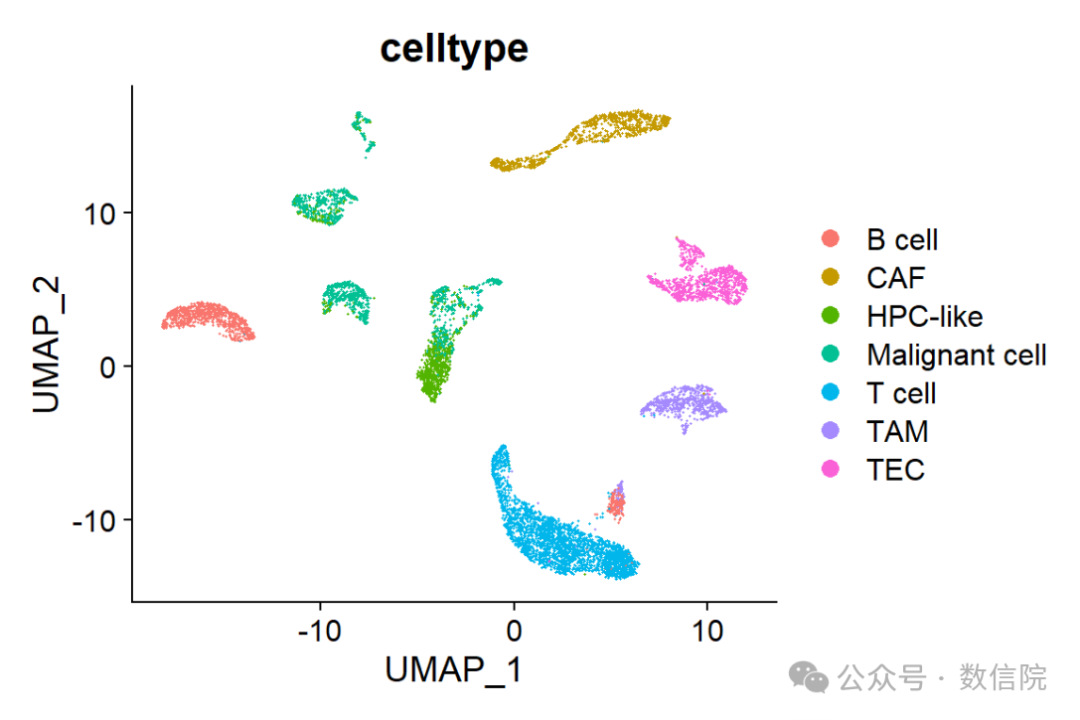
umap图
创建scAB对象并选择k参数scAB_data <- create_scAB(sc_dataset,bulk_dataset,phenotype)K <- select_K(scAB_data) #选4,函数select_K可以帮助选择k,当重构误差下降的趋势变缓时,k是一个合适的值。运行scAB模型scAB_result <- scAB(Object=scAB_data, K=K)sc_dataset <- findSubset(sc_dataset, scAB_Object = scAB_result, tred = 2)可视化与bulk表型高度相关的单细胞UMAP_scAB <- DimPlot(sc_dataset,group.by="scAB_select",cols=c("#80b1d3","red"),pt.size=0.001,order=c("scAB+ cells","Other cells"))patchwork::wrap_plots(plots = list(UMAP_celltype,UMAP_scAB), ncol = 2)

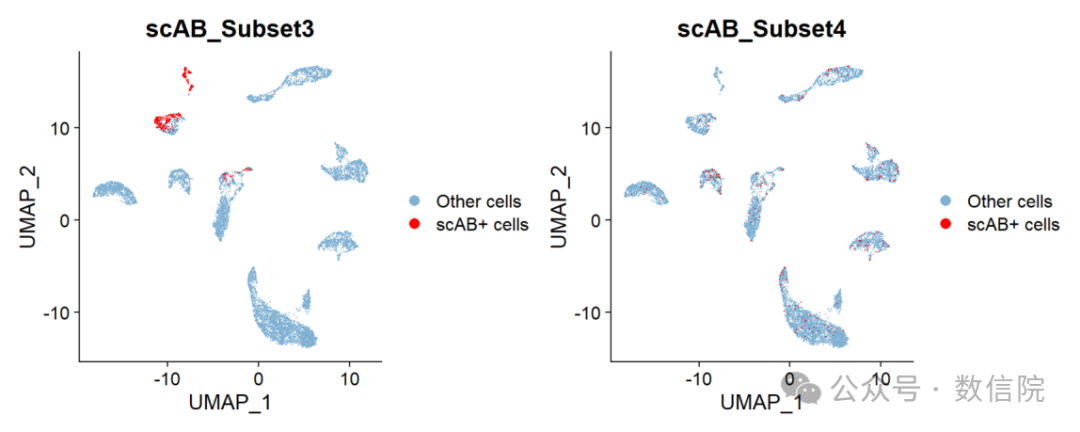
可视化单细胞的不同模块UMAP_subset3 <- DimPlot(sc_dataset,group.by="scAB_Subset3",cols=c("#80b1d3","red"),pt.size=0.001,order=c("scAB+ cells","Other cells"))UMAP_subset5 <- DimPlot(sc_dataset,group.by="scAB_Subset4",cols=c("#80b1d3","red"),pt.size=0.001,order=c("scAB+ cells","Other cells"))UMAP_subset <- patchwork::wrap_plots(plots = list(UMAP_subset3,UMAP_subset5), ncol = 2)UMAP_subset
识别与bulk表型相关的特征基因markers <- FindMarkers(sc_dataset, ident.1 = "scAB+ cells", group.by = 'scAB_select', logfc.threshold = 1)markers <- markers[which(markers$p_val_adj<0.05),]head(markers)#> p_val avg_log2FC pct.1 pct.2 p_val_adj#> CFHR1 0 1.690565 0.276 0.014 0#> TF 0 2.830617 0.420 0.045 0#> UGT2B4 0 1.033856 0.268 0.014 0#> GC 0 2.271937 0.478 0.069 0#> FGB 0 2.327561 0.427 0.053 0#> FGL1 0 2.114117 0.423 0.049 0
高认可度算法2——Scissor
发表于生信顶刊NBT[Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data],文章Fig1也清晰展示了算法的核心思想,输入为scRNA数据和bulkRNA数据,然后通过构建[细胞相似性网络]的操作,输出为每个单细胞和bulk表型相关的标签,最后就可以基于表型标签进行下游分析——分群/通路富集/生存分析等。
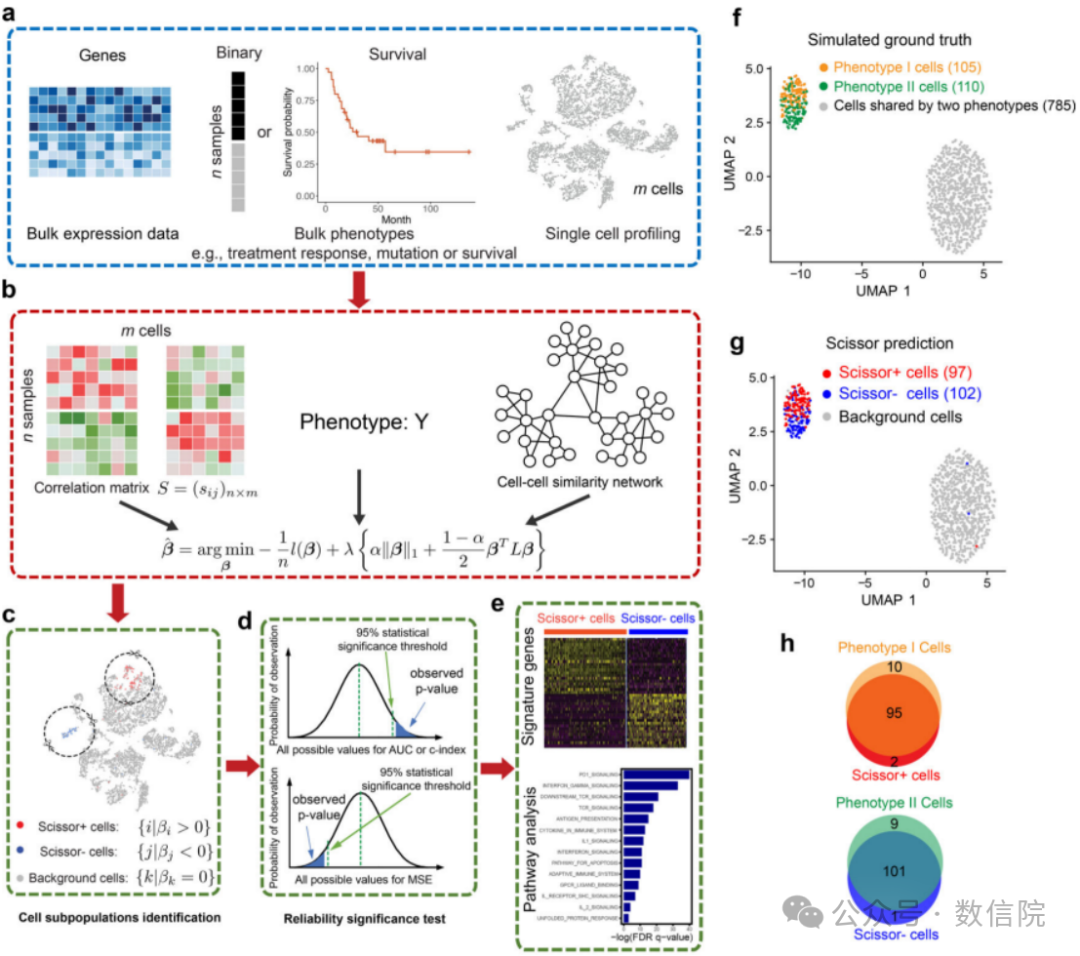
Fiure1
Scissor的代码实现——数信院共享服务器已经预装,上号即用
安装与数据加载
devtools::install_github('sunduanchen/Scissor')加载包和测试数据location <- "https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/"load(url(paste0(location, 'scRNA-seq.RData')))dim(sc_dataset)## [1] 33694 4102
单细胞数据预处理与分析——Scissor封装好的函数,默认支持Seurat v4对象
sc_dataset <- Seurat_preprocessing(sc_dataset, verbose = F)DimPlot(sc_dataset, reduction = 'umap', label = T, label.size = 10)

umap图
加载bulkRNA数据load(url(paste0(location, 'TCGA_LUAD_exp1.RData')))load(url(paste0(location, 'TCGA_LUAD_survival.RData')))dim(bulk_dataset)## [1] 56716 471head(bulk_survival)#TCGA_patient_barcode OS_time Status#TCGA-05-4249 1158 0#TCGA-05-4250 121 1#TCGA-05-4382 607 0#TCGA-05-4384 426 0#TCGA-05-4389 1369 0#TCGA-05-4390 1126 0phenotype <- bulk_survival[,2:3]colnames(phenotype) <- c("time", "status")head(phenotype)#Time status#1158 0#121 1#607 0#426 0#1369 0#1126 0
运行Scissor模型infos1 <- Scissor(bulk_dataset, sc_dataset, phenotype, alpha = 0.05, family = "cox", Save_file = 'Scissor_LUAD_survival.RData')## [1] "|**************************************************|"## [1] "Performing quality-check for the correlations"## [1] "The five-number summary of correlations:"## 0% 25% 50% 75% 100% ## 0.013191 0.196584 0.239458 0.298085 0.837278 ## [1] "|**************************************************|"## [1] "Perform cox regression on the given clinical outcomes:"## [1] "alpha = 0.05"## [1] "Scissor identified 201 Scissor+ cells and 4 Scissor- cells."## [1] "The percentage of selected cell is: 4.998%"## [1] "|**************************************************|"
这些选定的细胞占总数4102个细胞的5%。我们可以通过在Seurat对象sc_dataset中添加一个新的注释来可视化Scissor选择的细胞Scissor_select <- rep(0, ncol(sc_dataset))names(Scissor_select) <- colnames(sc_dataset)Scissor_select[infos1$Scissor_pos] <- 1Scissor_select[infos1$Scissor_neg] <- 2sc_dataset <- AddMetaData(sc_dataset, metadata = Scissor_select, col.name = "scissor")DimPlot(sc_dataset, reduction = 'umap', group.by = 'scissor', cols = c('grey','indianred1','royalblue'), pt.size = 1.2, order = c(2,1))

1代表Scissor阳性+细胞,2代表Scissor阴性-细胞
参数优化
在前面的示例代码中,我们将参数α设置为0.05 (alpha = 0.05),参数α代表平衡L1规范和基于网络的惩罚效果,较大的α倾向于强调L1规范以鼓励稀疏性,使得Scissor选择的细胞更少。
这里我们设置了一个新的百分比截断(cutoff = 0.03),当所选单元格的总百分比小于3%时,停止α搜索。
infos2 <- Scissor(bulk_dataset, sc_dataset, phenotype, alpha = NULL, cutoff = 0.03, family = "cox", Load_file = 'Scissor_LUAD_survival.RData')
为了避免在实际应用中任意选择alpha,我们建议根据所选细胞在总细胞中的百分比来搜索α。cut的默认值为0.2,即Scissor选择的细胞数量不应超过总细胞的20%。此外,用户可以设置自定义的α序列和百分比cut,以实现不同的目标。例如,我们可以这样运行Scissor:
infos3 <- Scissor(bulk_dataset, sc_dataset, phenotype, alpha = seq(1,10,2)/1000, cutoff = 0.2,family = "cox", Load_file = 'Scissor_LUAD_survival.RData')## [1] "alpha = 0.001"## [1] "Scissor identified 1666 Scissor+ cells and 1870 Scissor- cells."## [1] "The percentage of selected cell is: 86.202%"## ## [1] "alpha = 0.003"## [1] "Scissor identified 1262 Scissor+ cells and 37 Scissor- cells."## [1] "The percentage of selected cell is: 31.667%"## ## [1] "alpha = 0.005"## [1] "Scissor identified 889 Scissor+ cells and 28 Scissor- cells."## [1] "The percentage of selected cell is: 22.355%"## ## [1] "alpha = 0.007"## [1] "Scissor identified 729 Scissor+ cells and 24 Scissor- cells."## [1] "The percentage of selected cell is: 18.357%"## [1] "|**************************************************|"
scAB和Scissor用谁?
整体测试下来,我们认为scAB和Scissor的差距并不显著,但是Scissor的运行速度比scAB快很多,而且随着单细胞数据量的增加,其速度上升的更大。[然而Scissor是发表在生信顶刊NBT上的,可能是算法优化后面没有维护了]最后,如果你的数据量大,用Scissor即可~