说明:本文华算科技主要介绍化学键与杂化轨道在材料计算中的电子结构含义、常用图谱证据和材料功能判断方式。
化学键的基础概念在计算里对应什么变化
结构图里画出的 C-C、M-O 或 M-N 线,只是把相邻原子标出来。DFT 真正计算的是原子核势场中的电子密度和 Kohn-Sham 态;两个原子靠近后,哪些态得到占据、电子密度往哪里堆、总能降了多少,才决定这条线有没有成键含义。化学键在计算里先表现为电子密度和能量的重新分配,结构线条只是在结果图中把这种分配画得更直观。
在材料模型中,共价键、离子键、金属键和配位键更像几种极限情形:共价相互作用依赖定向轨道重叠,离子相互作用依赖电荷偏移和库仑吸引,金属键把电子态铺展到许多原子上,配位键常由配体孤对电子和金属空轨道参与。真实晶体很少完全符合某一个极限;一个氧化物表面、一个金属单原子位点、一个硫掺杂碳缺陷,通常带有局域重叠和电荷偏移两种特征。键型判断应依赖电荷密度、局域结构和轨道投影,单个键长或形式价态撑不起完整判断。
化学键
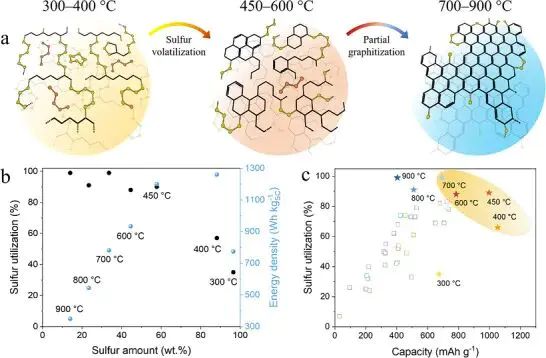
图1. 硫-碳材料热缩合过程中,碳骨架从 sp3 富集结构逐步转向 sp2 共轭结构,并伴随硫含量和硫利用率变化。
sp、sp2 和 sp3 常作为碳材料标签,图 1 里的硫-碳材料提醒我们,标签背后是局域配位和共轭程度在变。低温缩合阶段保留更多 sp3 富集碳链,升温后碳骨架逐步转向 sp2 共轭网络,硫物种的束缚位置、导电路径和离子可接近的位点都会跟着移动。杂化标签只有绑定局域结构才有判读价值,孤立写成“某材料是 sp2”很难解释电子结构差异。
优化结构时,程序并不知道哪条线应该保留。力把原子推到能量更低的位置,电子密度在新的核构型下重新排布;若两个原子之间出现电子积累,相关轨道在能量轴上分裂出占据的成键态,反键态占据较低,结构图里的那条线才有电子结构支撑。化学键是能量降低后的电子结构结果,并非两个元素符号连线后自动成立的静态分类。
周期晶体中的成键问题还处在 Bloch 态和局域投影的共同约束下:价带深处可能藏着稳定骨架,费米能级附近可能是离域金属态,缺陷又会在带隙或带边引入局域态。读一个材料的“化学键”时,只盯着优化后的距离容易漏掉能量区间差异;键长、电荷密度和能带投影各自回答不同问题,几何、空间分布和能量位置要互相对上。
杂化轨道为什么会随局域结构改写
杂化轨道最早说的是同一原子上 s、p 或 d 成分重新组合,形成更适合成键方向的轨道。到了固体和表面模型里,“轨道杂化”常指不同原子、不同能量区间、不同空间取向的电子态发生混合。能量接近、对称匹配和空间重叠一起出现时,能谱里才会分出成键态和反键态。
表面、缺陷和界面会改变原子的近邻数、键角和未饱和轨道。图 2 里,graphdiyne 的 sp 碳与 ZnS 纳米簇接触后,C-S-Zn 界面键把碳骨架、硫位点和 ZnS 簇的电子态连到同一个局域结构里;ZnS 放在 graphene 上时,界面更接近较远的范德华接触。局域键合方式已经换成另一类相互作用,后续 Hg0 吸附便会选到不同位点。
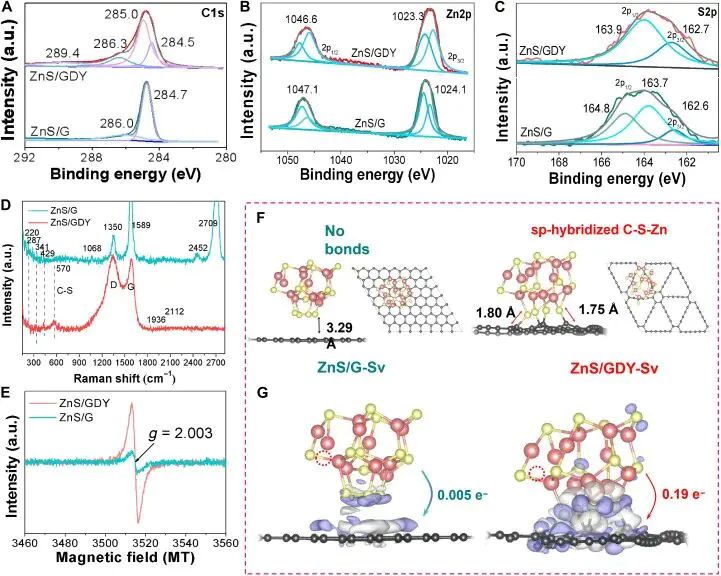
图2. ZnS/GDY 的 XPS、Raman、EPR、DFT 结构模型和差分电荷密度图,右侧模型给出 sp 杂化碳参与的 C-S-Zn 界面键。
杂化也不要求整块材料都改成某一种键型。Cr3TMSe6 这类层状硒化物里,TM-Se 八面体和 Cr-Se 网络的近邻环境不同,替位元素的 3d 或 5d 态会改变金属-硒之间的离域程度。图 3 的 ELF 截面把这种差别画在实空间里:某些区域电子更局域,另一些区域更接近离域背景。同一晶胞内可以并存局域电子、离域 d 态和混合离子-共价键,单一键型标签会把这些差别抹平。
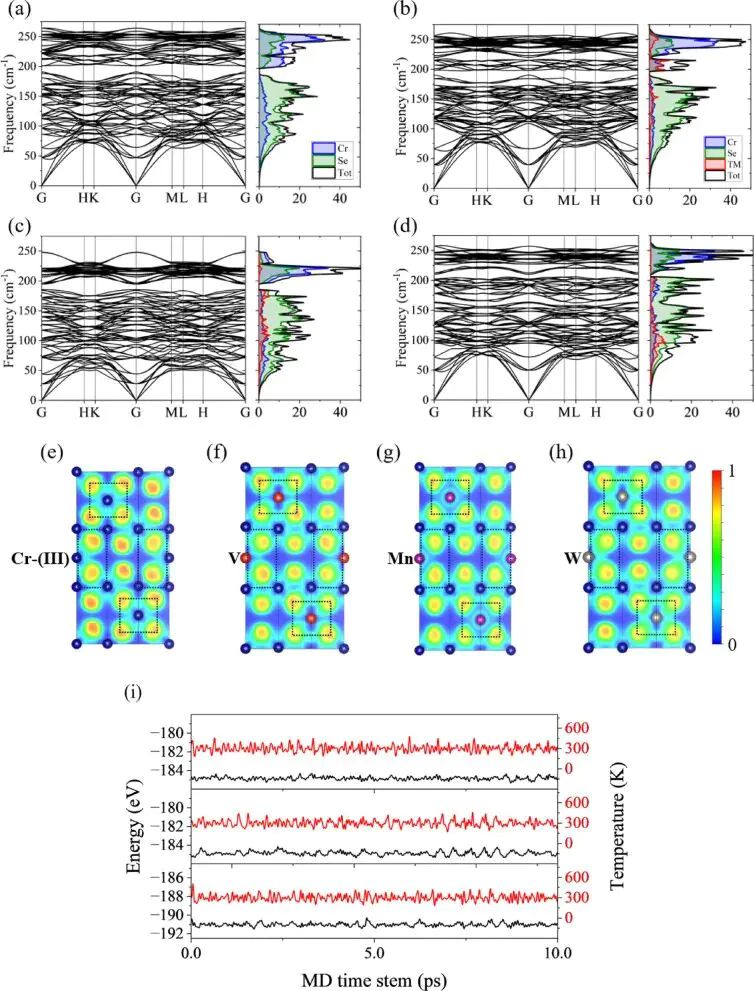
图3. Cr2Se3 与 Cr3TMSe6 的声子谱、ELF 截面和 AIMD 曲线,ELF 色标用于比较局域键合环境。
同一组原子在深价带、费米能级附近和未占据区间里,常常对应不同电子过程。深价带里的态多半对应稳定骨架,费米能级附近的态会牵动输运、磁响应或吸附响应,未占据态更多参与激发或受电子过程。谈杂化时必须写明能量参照和投影对象,例如金属 d 态与配体 p 态、吸附物 s 态与表面 p/d 态,或者缺陷态与带边态之间的混合。
octahedral 场中的 dz2、dxz/dyz 和 dxy/dx2-y2 面向不同配体方向,轨道方向本身就会改变杂化读法。层状材料的面内键和层间键会选择不同轨道成分。同一个 d 元素若只写成 d 态参与杂化,会丢掉轨道分量和晶体场环境对峰位与键合方向的限制。
哪些图谱能区分成键、反键与轨道杂化
在吸附或掺杂的 PDOS 图里,最常见的误读是把峰重叠直接写成“键更强”。两个轨道在同一能量区间有峰,并且峰位随结构变化移动,确实说明相关态开始混合;但能量轴上的重叠还没有给出相位关系,也没有给出反键态填了多少电子。成键还是反键由相位、占据和对应原子对的哈密顿布居贡献决定,PDOS 需要与 COHP、键长和电荷密度共同约束。
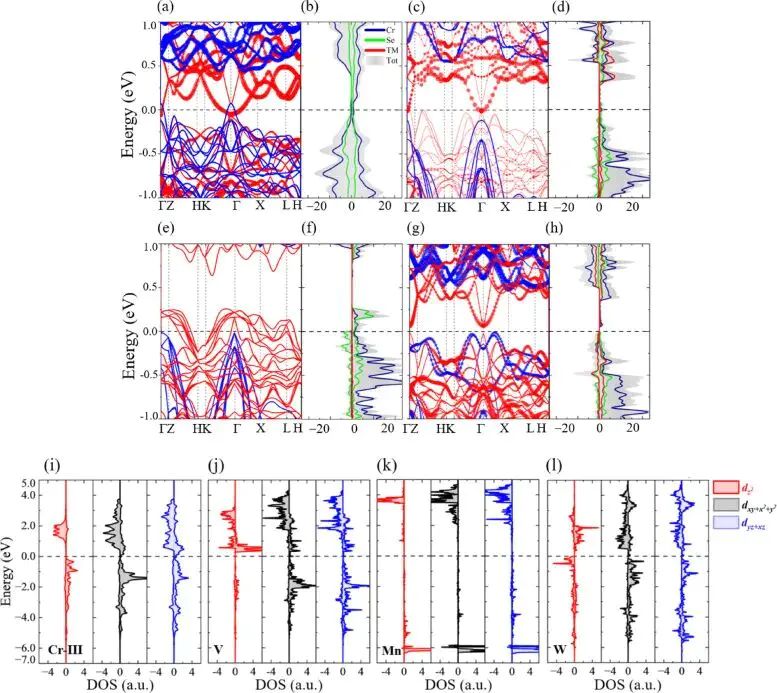
图4. Cr3TMSe6 的自旋能带、元素投影态密度和轨道分辨 PDOS,气泡大小标示 3d-3d 或 3d-5d 轨道杂化贡献。
ELF 接近 1 的区域表示电子更局域,靠近 0.5 的区域更接近离域电子气;差分电荷密度比较成键前后电子密度增减,能看出电子在界面、缺陷或吸附物附近的积累和耗尽。ELF 看局域程度,差分电荷密度看重排方向,键能数值仍要来自能量差、成键布居或积分后的相互作用指标。
COHP、ICOHP、COOP 或键级类指标适合处理“这一对原子到底在成键还是反键”这样的窄问题。负的 COHP 常对应成键贡献,正的 COHP 常对应反键贡献,符号约定要按软件输出核对;积分值把某个能量范围内的贡献压成数值。指定原子对、积分范围和费米能级位置写清后,COHP 才能和 PDOS、ELF 互相校验。
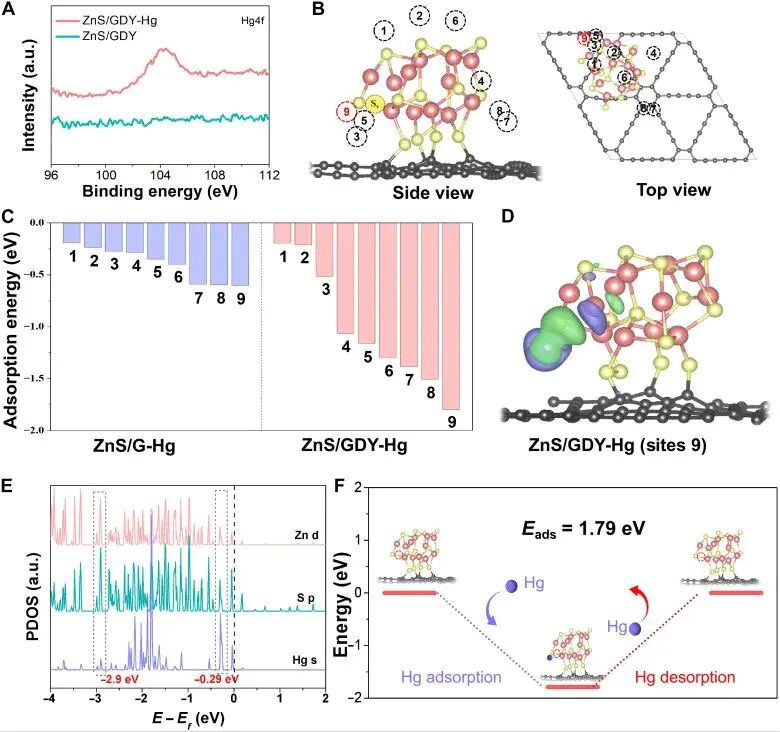
图5. Hg0 吸附在 ZnS/GDY 后的构型、电荷密度差、PDOS 和吸附/脱附能图,PDOS 区分 Zn d、S p 与 Hg s 态贡献。
Bader、Hirshfeld 或 Mulliken 分区给出原子盆地内的电子数变化,适合补足差分电荷密度的图像感。它们回答的是积分电子数如何变,不会自动给出形式价态或键能。成键分析中,布居数更适合与键长、COHP 积分和 PDOS 峰位相互约束。
Hg0 与 ZnS/GDY 中 S 位点相互作用时,S p 态和 Hg s 态在相关能量区间配合吸附能,差分电荷密度又显示界面附近电子重排,化学吸附图像才站得住。单独的峰重叠不宜写成键强排序,它必须依赖位点类型、吸附构型和电子占据。
怎样把化学键描述用于材料功能判断
化学键能解释材料功能,但要写到可计算的变化上。图 6 里的 Fe-N5 位点,配位数改变了 Fe 3d 态位置,氧中间体吸附自由能随之变化;电池正极讨论 M-O 键时,读者关心氧空穴、结构畸变和氧氧化还原;二维磁体讨论金属-配体杂化时,交换路径和局域磁矩才是落点。化学键描述应连接结构、电子态和目标响应,并保留模型条件。
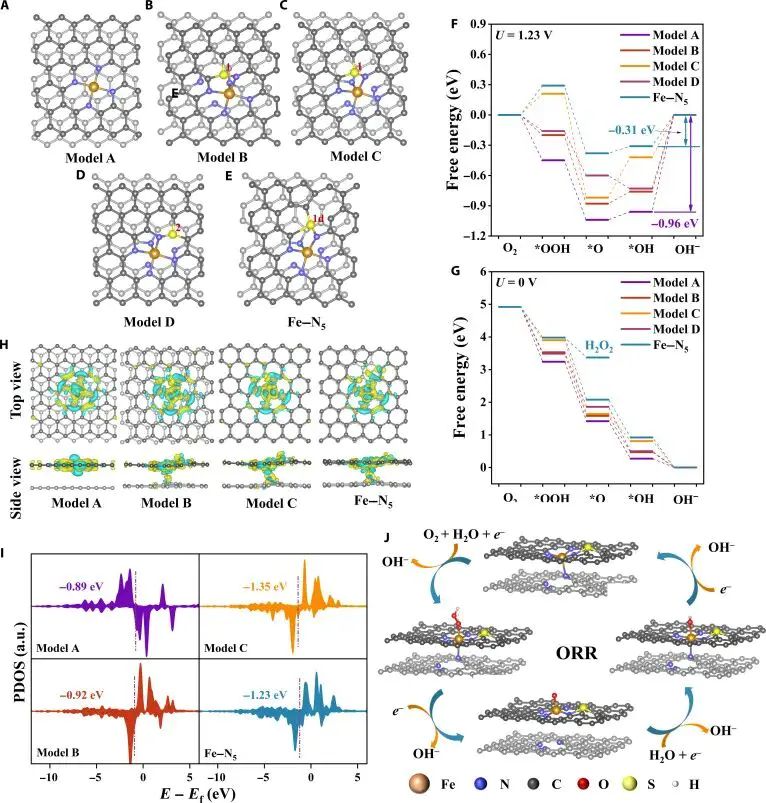
图6. Fe-N5 位点的 DFT 模型、自由能、电荷密度差、Fe 3d PDOS 和 ORR 路径,局域配位改变 Fe 位点电子结构。
硫掺杂 graphene 的 basal-plane 缺陷更能说明“位点”这个词的重量。图 7 中,S、O、C 周围的配位和羟基修饰会形成 sp3-like 碳位点、极性键和局域磁矩;这些局域状态改变吸附中间体的取向和自由能。缺陷成键图像要写成位点级问题,把整片碳材料归为一个均匀表面,会掩盖位点之间的配位差异。
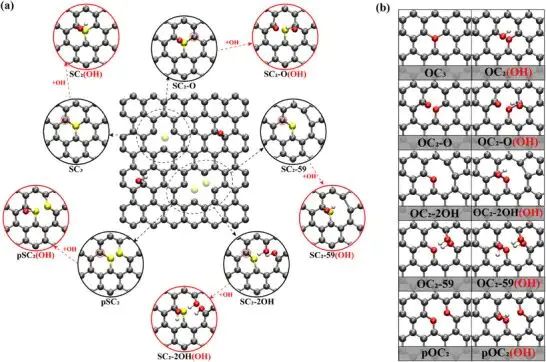
图7. 硫掺杂 graphene 和含氧 basal-plane 缺陷构型,红圈标出羟基修饰后形成的局域 sp3-like 碳环境。
成键越强,功能越好,这句话在材料计算里经常出错。金属-吸附物键过弱,反应物不容易活化;过强,又可能把产物困在表面。p-d 杂化增强时,层间交换可能上升,带隙和载流子有效质量随之改变。金属 d 态更离域时,导电响应可能变强,局域磁矩却可能下降。成键强弱、反键占据和材料功能之间没有固定单调关系,判断要跟吸附自由能、交换路径或输运指标连在一起。
催化吸附要看 Eads、ΔG 和关键中间体的 PDOS/COHP;磁耦合要看交换路径、局域磁矩和 p-d 杂化;电池正极要看 M-O 共价程度、氧空穴和结构畸变。同一套成键图谱用于不同研究对象后,参考态、能量窗口和读图重点都会改变。把所有问题都写成“轨道杂化增强”,会把真正的判断对象藏起来。
写材料计算结果时,化学键与杂化轨道适合承担电子结构解释。结构优化给出键长和配位,PDOS 给出轨道来源,ELF 与差分电荷密度给出实空间分布,COHP 或键级指标给出指定原子对的成键/反键贡献,吸附能、形成能、NEB 能垒和自由能图再把这种电子结构差异接到材料功能。判断范围停在给定材料状态、能量窗口和图谱信号上,例如 C-S-Zn 界面键、S p-Hg s 杂化峰、Fe 3d 态移动或 Cr-Se ELF 局域区域。