编者按
代谢相关脂肪性肝病(MASLD)及其更严重的炎症形式——代谢相关脂肪性肝炎(MASH),已成为全球性的重大健康挑战。然而,针对此病的有效药物治疗手段仍然有限。G蛋白偶联受体75(GPR75)是GPCR家族中的一个孤儿受体。此前的人类遗传学研究表明,GPR75的功能缺失变异与较低的体脂率和身体质量指数(BMI)相关。在动物模型中,全身性敲除Gpr75的小鼠能够抵抗高脂饮食诱导的肥胖,并改善胰岛素敏感性。然而,这些研究多集中于GPR75通过调控食物摄入来影响全身代谢,其在肝脏中是否直接调控脂质积累和MASH进展的机制尚不明确。发表在Hepatology的一项研究首次深入揭示了肝细胞特异性GPR75在MASH发生发展中的关键作用及其分子机制,并提出了以GPR75为靶点的治疗新策略。
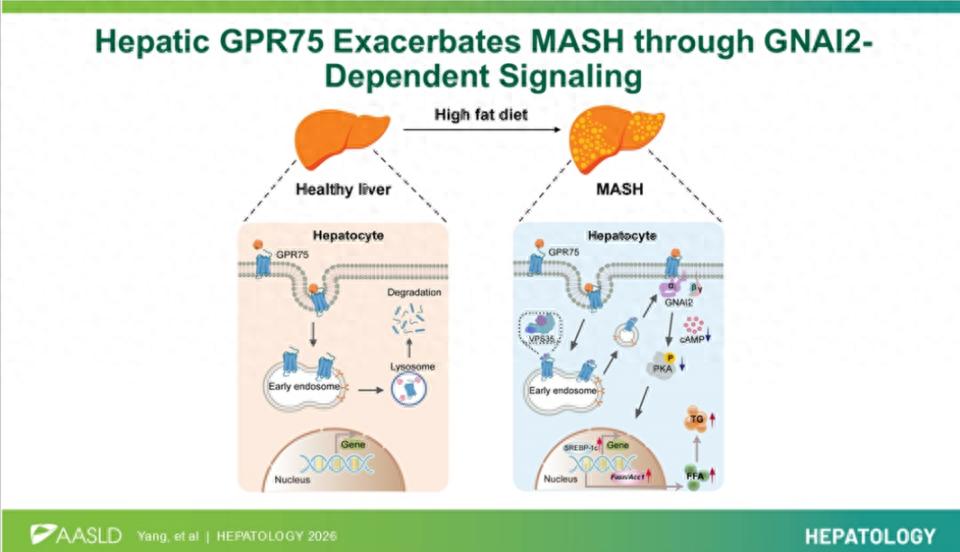
文章图形摘要
MASH是代谢综合征在肝脏的表现,其特征是肝细胞脂肪变性、炎症、气球样变和纤维化。肝脏脂质代谢的失调是核心致病环节。GPR75在健康个体的肝脏中表达量并不高,但全外显子组测序数据显示,其基因变异与较低的肝脂肪变性风险相关。有趣的是,促进瘦身的基因变异有时会矛盾地增加脂肪肝风险,这提示我们需要更精细地理解特定基因在特定组织(如肝脏)中的功能。此外,尽管已有研究发现趋化因子CCL5和二十羟二十碳四烯酸(20-HETE)可能是GPR75的内源性配体,但GPR75在MASH中的直接作用机制仍为明晰。
基于此,该研究旨在明确GPR75在肝脏MASLD/MASH进展中的具体功能,并阐明其下游信号通路,以评估其作为治疗靶点的潜力。
核心发现01
GPR75蛋白在MASH肝脏中特异性上调
研究人员首先在人类和小鼠样本中验证了GPR75的表达情况。通过对公共基因表达数据库(GEO)的分析以及定量PCR检测,他们发现无论是在MASH患者还是饮食诱导的MASH小鼠肝脏中,GPR75的mRNA水平与正常对照相比均无显著差异。然而,蛋白质水平的检测却呈现出截然不同的结果:免疫组化染色显示,在MASH及肝纤维化患者的肝组织中,GPR75蛋白水平显著增加;蛋白质印迹和免疫荧光实验进一步证实,在高脂饮食(HFD)或胆碱缺乏高脂饮食(CDAHFD)诱导的MASH小鼠肝脏中,GPR75蛋白同样被显著上调。这一发现揭示了GPR75在MASH状态下存在转录后水平的调控,其蛋白稳定性或运输过程可能发生了改变。
02
肝细胞GPR75缺失缓解MASH,而过表达则加重MASH
为了探究GPR75上调的功能意义,研究团队构建了肝细胞特异性敲低、敲除及过表达Gpr75的小鼠模型。
敲低/敲除实验:通过腺相关病毒(AAV8)介导的肝细胞特异性敲低Gpr75,或利用Cre-loxP系统构建肝细胞特异性敲除(Gpr75hko)小鼠,并用CDAHFD或HFD喂养。结果表明,降低肝细胞GPR75表达能够显著减轻小鼠的肝脏重量/体重比、肝脂肪变性(HE和油红O染色显示脂滴减少)、肝脏甘油三酯(TG)和游离脂肪酸(NEFA)含量。同时,肝损伤标志物(血清ALT/AST)、炎症因子(如TNF-α, IL-1β, IL-6)和纤维化标志物(如α-SMA, COL1A1)的表达也明显下降。重要的是,肝细胞特异性敲除Gpr75还能改善HFD小鼠的全身糖代谢和胰岛素敏感性。
过表达实验:相反,通过AAV8介导的肝细胞特异性过表达Gpr75,则加重了饮食诱导的MASH表型。过表达小鼠表现出更严重的肝脂肪变性、炎症细胞浸润、胶原沉积(纤维化)、肝损伤以及脂质合成相关基因的上调。
这些功能获得和功能缺失实验强有力地证明了肝细胞GPR75是驱动MASH进展的关键促进因子,其表达水平与疾病严重程度正相关。
03
GPR75通过偶联GNAI2抑制cAMP-PKA通路,促进脂质合成
作为GPCR,GPR75需要偶联G蛋白来传递信号。为了寻找其下游效应分子,研究者对对照和GPR75敲低小鼠的肝脏进行了蛋白质组学分析。基因富集分析提示GPR75与GTP结合等通路相关。进一步的火山图分析发现,抑制性G蛋白亚基Gαi2(由Gnai2编码)在GPR75敲低后显著减少。后续实验验证了GNAI2的mRNA和蛋白水平均受GPR75表达量的调控。
相互作用验证:免疫共沉淀(Co-IP)实验在棕榈酸处理的HepG2细胞中证实了GPR75与GNAI2之间存在直接的蛋白相互作用。免疫荧光染色也显示两者在MASH小鼠肝脏中存在共定位。
信号通路解析:G蛋白Gαi亚家族的主要功能是抑制腺苷酸环化酶(AC)的活性,从而降低细胞内第二信使环磷酸腺苷(cAMP)的水平,进而抑制蛋白激酶A(PKA)的活化。本研究提出并验证了以下通路:GPR75通过偶联并可能激活GNAI2,抑制了AC-cAMP-PKA信号通路。PKA活性的降低,导致其下游底物——固醇调节元件结合蛋白1c(SREBP-1c)的磷酸化受到抑制。
关键转录因子SREBP-1c的作用:SREBP-1c是调控肝脏脂肪从头合成(DNL)的关键核转录因子。PKA通常通过磷酸化SREBP-1c或其上游调控因子(如肝脏X受体α, LXRα)来抑制其活性和成熟,从而减少脂质合成。本研究发现,GPR75过表达抑制PKA磷酸化,增加了SREBP-1c(特别是其成熟的核形式)的蛋白水平,进而上调了Acc1, Fasn, Scd1等脂质合成基因的表达,最终导致肝细胞脂质积累。相反,敲低GPR75或敲低GNAI2都能激活PKA,降低SREBP-1c水平,缓解脂质堆积。体内实验也证明,在GPR75过表达的小鼠中,同时敲低Gnai2可以逆转其加重的肝脂肪变性、炎症和纤维化表型。
04
上游调控:VPS35通过循环运输维持GPR75的膜蛋白稳定性
一个关键问题是:为何MASH时GPR75蛋白水平升高而mRNA不变?研究者从蛋白降解和运输角度进行了探索。他们发现,在MASH状态下,GPR75的降解速度减慢。通过免疫沉淀联合质谱(IP-MS)分析寻找与GPR75相互作用的蛋白,并将目标锁定在逆运复合体的核心组分VPS35上。VPS35已知负责将膜蛋白从内体循环运输至细胞膜,而非运往溶酶体降解。
实验证据:Co-IP和免疫荧光证实VPS35与GPR75在肝细胞内相互作用并共定位。机制上,VPS35通过将GPR75回收到肝细胞膜上,减少了其被溶酶体降解的机会,从而在转录后水平稳定并上调了GPR75的蛋白表达。沉默VPS35会加速GPR75的降解,减弱棕榈酸诱导的肝细胞脂质积累。
体内功能:在CDAHFD喂养的小鼠中,敲低肝脏Vps35能够改善MASH表型,包括减轻脂肪变性、炎症和纤维化。重要的是,Vps35敲低导致了GPR75蛋白水平的下降,并激活了PKA磷酸化,其效果与直接敲低Gpr75类似。临床数据分析也显示,MASH患者肝脏中VPS35的表达上调。
因此,VPS35-GPR75轴构成了一个在MASH中放大致病信号的正反馈环路:疾病状态上调VPS35,VPS35稳定并增加膜上的GPR75,GPR75再通过GNAI2-cAMP-PKA-SREBP-1c通路促进脂质合成与炎症,加剧MASH。
05
治疗转化:靶向GPR75的化合物筛选
基于上述机制,研究团队以GPR75为靶点,利用计算机辅助分子对接技术筛选具有高亲和力的化合物。其中,化合物X201415(CAS 477252-30-5)在细胞热转移实验(CETSA)中显示能与GPR75结合并提高其热稳定性。在HepG2细胞中,X201415能以剂量依赖的方式抑制棕榈酸诱导的脂滴积累,且该保护作用在GPR75被敲除后消失,证明其特异性作用于GPR75。
在CDAHFD诱导的MASH小鼠模型中,口服给予X201415(20 mg/kg/天)进行治疗。结果显示,治疗显著降低了小鼠的肝脏重量、减轻了肝脂肪变性和纤维化程度,降低了肝脏TG含量,并下调了炎症和纤维化相关基因的表达。在机制上,X201415处理激活了肝脏中的cAMP-PKA通路,并降低了SREBP-1c蛋白水平。
小 结该研究将GPR75确立为肝脏脂质代谢和MASH进展的一个新型关键调节因子,完整描绘了从VPS35介导的膜蛋白稳定,到GPR75-GNAI2偶联,最终通过经典cAMP-PKA-SREBP-1c轴调控脂质合成的完整信号链条。它为解决当前MASLD/MASH领域药物治疗匮乏的困境提供了新的思路和实验基础。未来的研究可进一步探索GPR75特异性拮抗剂的优化、其在不同细胞类型中的作用,以及该通路在人类MASH患者中的临床相关性,从而加速相关疗法向临床应用的转化。
参考文献:Yang X, Yang N, Cheng Z, et al. Hepatic GPR75 exacerbates MASH through GNAI2-Dependent signaling. Hepatology. Published online February 3, 2026. doi:10.1097/HEP.0000000000001706