说明:本文华算科技主要介绍第一性原理在材料计算中的物理起点、计算对象、常见实现和适用范围。
第一性原理这个基础概念从哪里开始
第一性原理的“第一”,指材料描述从更基础的物理对象出发:原子核的种类和位置、电子数、电荷相互作用以及量子力学方程。给定元素组成和结构模型后,体系的总能量、电子密度、受力和能级不靠某个目标性质的经验公式直接指定,而由电子结构计算给出。计算起点决定结果含义,经验拟合退到较少的位置,材料性质从电子和原子核之间的相互作用中导出。
在材料计算语境里,ab initio、first-principles 和第一性原理常被放在相近位置。它们通常指向同一类思路:先建立体系的哈密顿量或能量泛函,再用可求解的近似形式得到基态电子结构。DFT 是最常见实现,第一性原理方法还包括其他量子多体求解路线,很多日常材料计算把“第一性原理”默认指向 DFT;高精度量子化学、量子蒙特卡罗、GW 或 BSE 同样可以承担第一性原理计算角色。
元素种类、原子坐标、电荷态、自旋态、周期性约束、真空层、赝势、基组和交换相关泛函,都是第一性原理计算的明确输入。它与经验模型的区别在于,计算并未预设目标材料的带隙或某个表面对 H* 的吸附能;这些性质由电子结构求解和后处理返回。目标性质来自计算输出,而非经验答案输入。

图1. AiiDA 的工作流、计算节点和数据节点示意,展示计算结果如何追溯到输入数据与中间过程。DOI:10.1038/s41597-020-00638-4
在可追溯计算图中,输入数据、计算节点、工作流节点和输出数据之间保留完整关系。第一性原理结果的关键内容是可复查的物理量,同时包括计算来源和比较基准。一个能量、密度或图谱若能追到具体结构模型与计算过程,它的物理含义才有明确对象。
材料结构怎样变成能量、力和电子密度
材料计算从结构模型开始。周期晶体给出晶胞和原子坐标,表面模型给出 slab、吸附物和真空层,缺陷模型给出超胞和缺失或替位原子。原子核的位置决定电子感受到的外势,电子在由原子核产生的外势中重新分布,体系总能随结构改变而改变。第一性原理计算对象是结构模型中的电子占据,材料名本身没有给出几何构型、约束条件和电荷状态。
Born-Oppenheimer 近似把快电子和慢原子核运动分开处理。对于一次静态电子结构计算,原子核位置先固定,电子达到基态后给出总能量和受力;结构弛豫再沿着力的方向更新原子坐标,直到残余力足够小。这样得到的平衡结构、键长、表面重构和吸附构型,来自同一套电子能量面。能量面和力把结构优化连接起来,力由总能量对原子位移的导数给出,来源仍是同一套电子能量面。
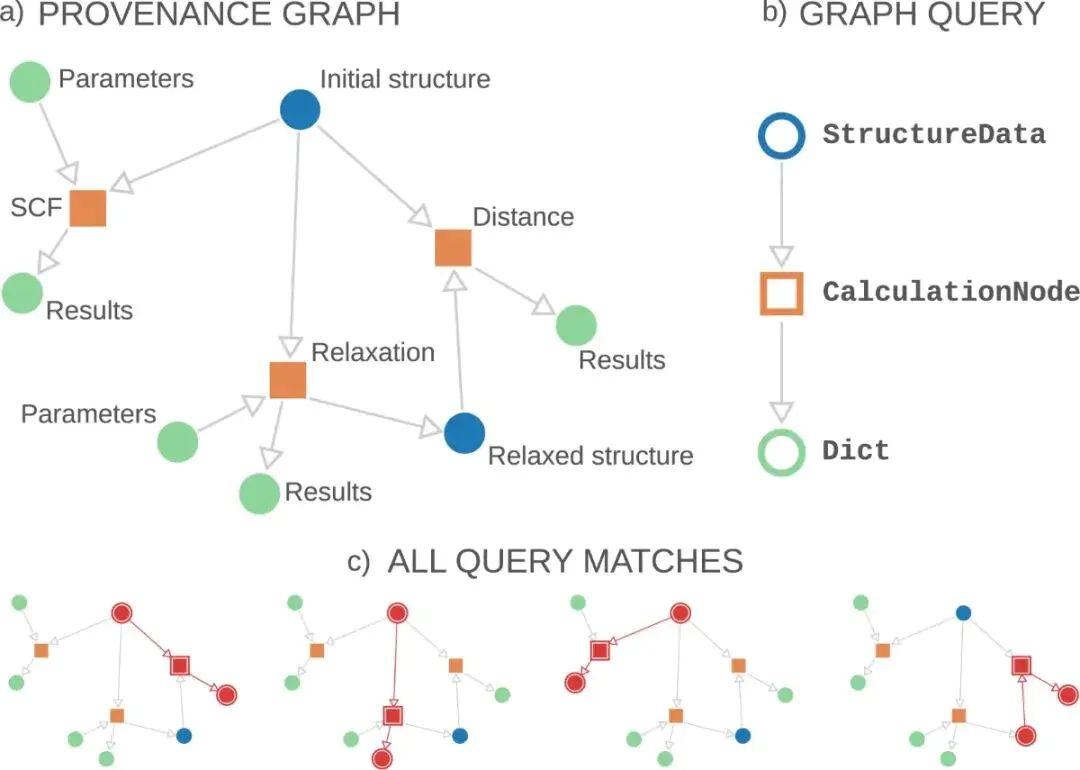
图2. 可追溯计算图中包含 DFT-SCF、结构弛豫、初始结构、弛豫结构和结果节点。DOI:10.1038/s41597-020-00638-4
真实研究里,第一性原理很少只产生一个数。一次吸附体系计算可能同时得到弛豫构型、Eads、电荷密度、DOS/PDOS、差分电荷密度和反应中间体的相对能量。不同输出对应不同问题:总能差比较热力学趋势,受力和应力约束结构稳定,电子密度描述空间电荷分布,能带和态密度描述可用电子态。所有输出若被压成同一种“性能好坏”判断,物理对象差别会被抹平。
SCF 自洽是很多 DFT 结果的共同计算内核。初始电子密度进入 Kohn-Sham 方程,得到新的电子态和电子密度,再反复更新到密度、能量或势场变化足够小。结构弛豫则在每一步调用电子结构计算获得力。初始结构、计算参数、SCF 结果、弛豫结构和后处理结果共同构成结果来源,不同材料或不同位点的比较也建立在这些来源信息之上。
为什么第一性原理结果仍会出现差异
第一性原理有基础物理起点,但实际求解离不开近似。电子-电子相互作用中最难处理的交换相关部分,在 DFT 中由 LDA、GGA、meta-GGA、混合泛函等不同形式描述;核心电子与价电子的分工,常由赝势或 PAW 数据集完成;周期体系还要选择平面波截断能、k 点采样、展宽方式和收敛标准。差异常常来自近似类型和数值设置,同一个材料在不同实现里可能得到不同数值。
AFLOW、Materials Project 和 OQMD 这类高通量数据库都属于第一性原理数据来源,形成能、体积、带隙、磁化等性质却会出现不同程度的分散。形成能和体积通常更一致,带隙和总磁化对泛函、U 值、自旋设置和赝势更敏感。第一性原理数据不会自动收敛到唯一标准答案,同一物理起点经过不同计算协议,会留下可比性差异。

图3. AFLOW、Materials Project 和 OQMD 数据库中形成能、体积、带隙和总磁化的计算差异分布。DOI:10.48550/arXiv.2007.01988
形成能属于能量差问题,在许多固体数据集中,相近泛函和一致参考态会给出较接近的数值;带隙涉及 Kohn-Sham 本征值和交换相关势,PBE 常低估半导体带隙,HSE 或 GW 会改变带边位置;磁性体系还对初始磁矩、自旋构型和局域 d/f 电子处理敏感。第一性原理结论对应的是具体物理量,数值越靠近强关联、开壳层或磁性问题,方法选择的影响越明显。
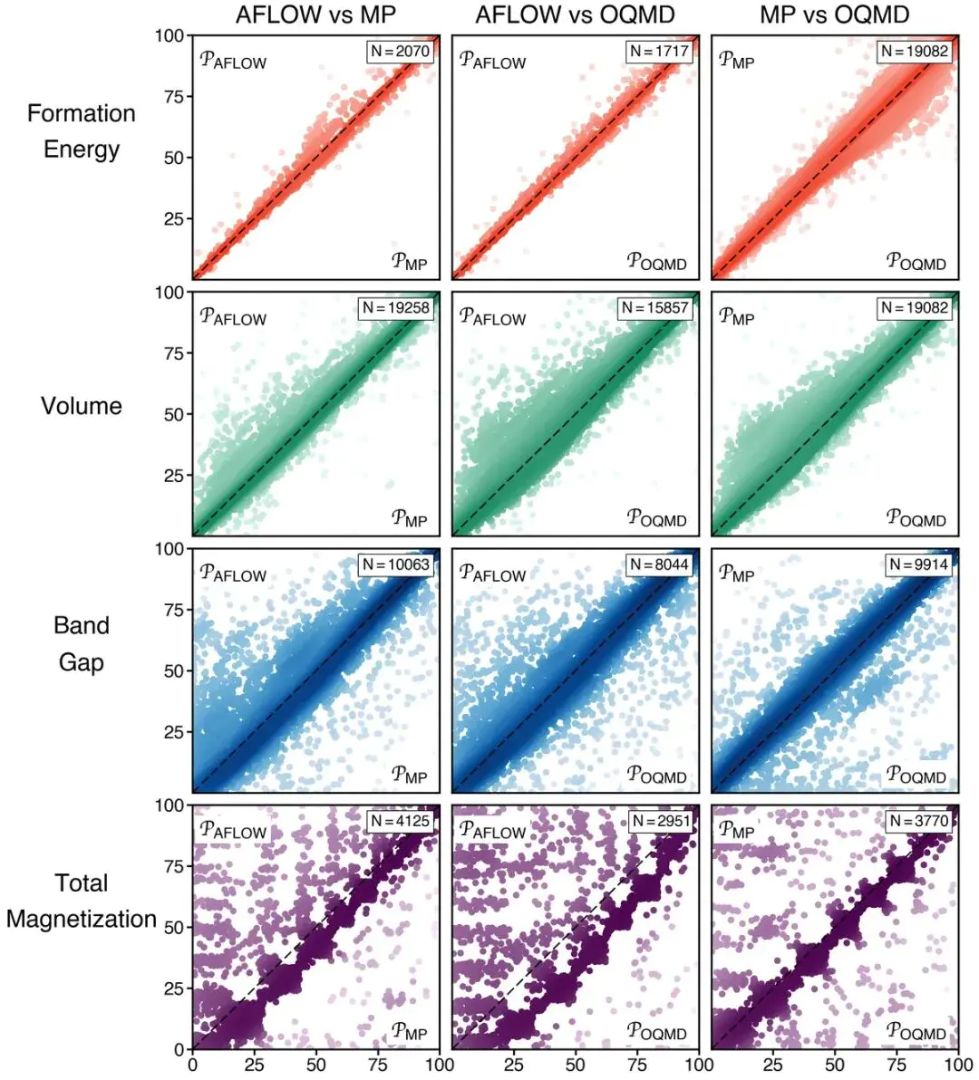
图4. 三个高通量 DFT 数据库中材料性质的百分位排名对比,形成能和体积相关性高于带隙和磁化。DOI:10.48550/arXiv.2007.01988
普通 sp 电子半导体、过渡金属氧化物、稀土化合物、强磁性体系和含重元素材料,对泛函、U 值、SOC、赝势半芯态和相对论处理的敏感程度存在明显差别。计算条件需要与材料电子结构特征匹配,同一套 DFT 设置可满足某类体系的筛选精度,在另一类体系中可能只能提供初筛排序。
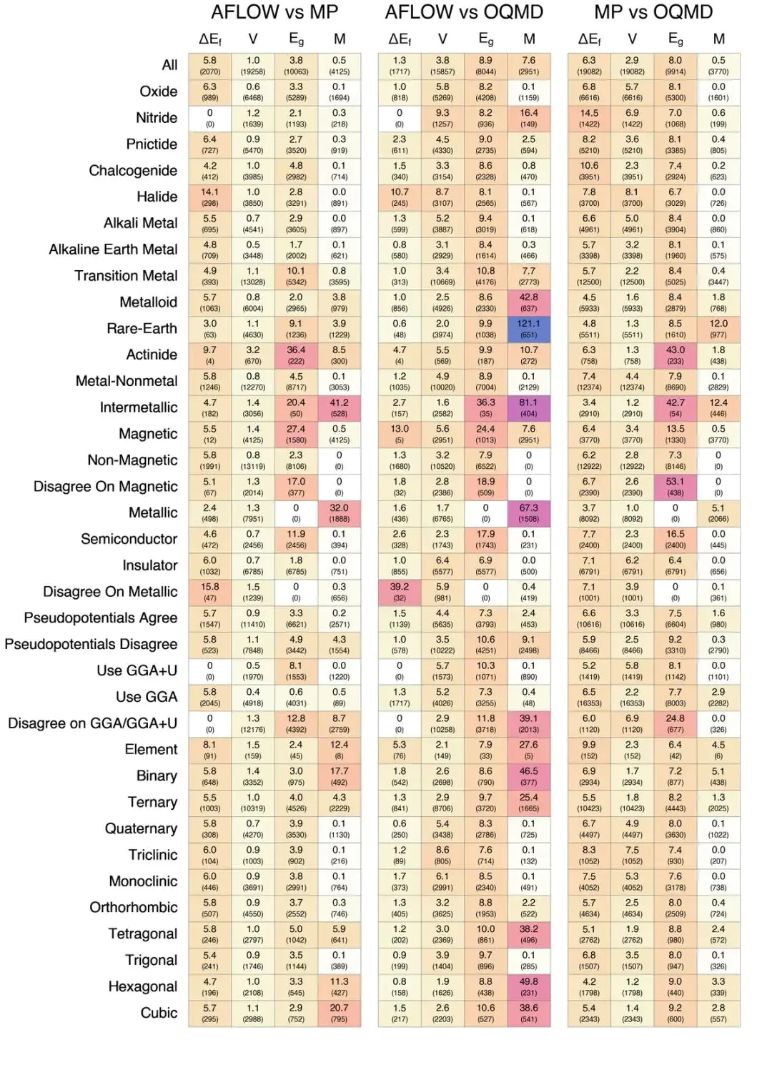
图5. 不同材料类别中,形成能、体积、带隙和总磁化在数据库之间呈现不同中位百分差异。DOI:10.48550/arXiv.2007.01988
按元素统计的形成能差异还指向一个来源:材料数据库中的数值差别不只来自“算法好坏”,还来自元素参考态、赝势定义、能量修正和可比样本分布。模型条件和近似设置共同限定结果含义,同一个“第一性原理”标签下,对应的结构设定和参考基准可能完全不同。
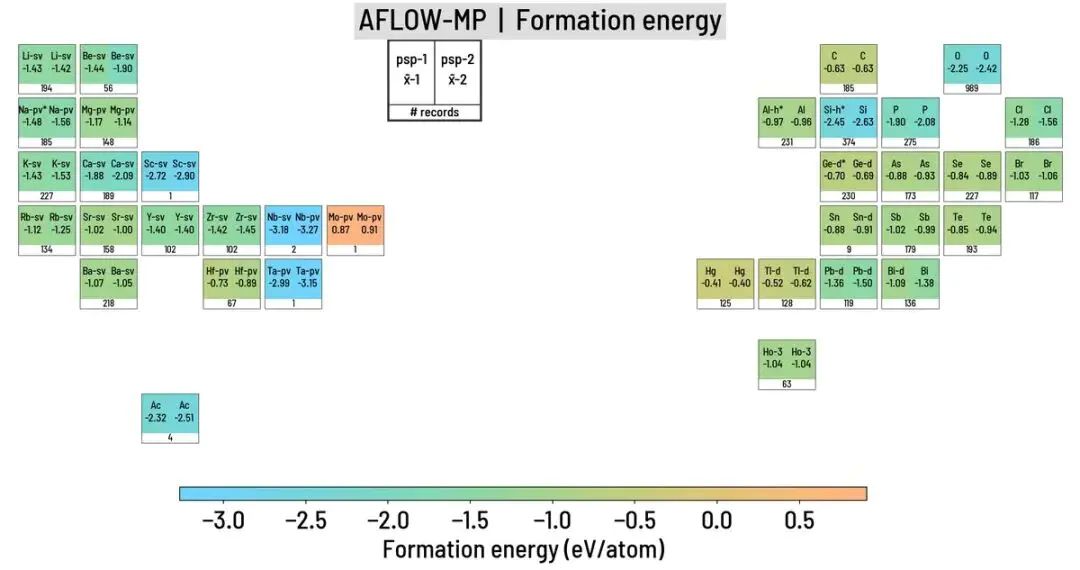
图6. AFLOW-MP 对比中,按元素统计的化合物形成能中位值和赝势标识。DOI:10.48550/arXiv.2007.01988
还有一类差异来自模型本身。slab 厚度会影响表面能和功函数,覆盖度会改变吸附能,缺陷电荷态会受费米能级和化学势约束,二维材料需要检查真空层和偶极修正,电化学界面还涉及溶剂、电位和双电层。材料体系越靠近表面、界面、缺陷和强关联电子,计算条件对结论适用范围的限制越强。
第一性原理结果到底能支持什么判断
第一性原理适合支持“在给定模型和计算条件下,某个结构、电子态或能量差怎样变化”的判断。它能比较不同构型的相对总能,给出原子受力和结构弛豫方向,分析费米能级附近的态密度,定位差分电荷密度中的电子累积和耗尽区域,也能为吸附、扩散、相稳定和反应路径提供能量基准。可比较性来自明确的比较对象和参考态,孤立数值只覆盖局部信息。
第一性原理给的是受模型限定的证据,吸附能、带隙和 Bader 电荷分别对应局部问题,材料性能很难由一个数值覆盖。吸附能较负只能说明该模型下相互作用较强,催化反应还涉及中间体转化、产物脱附和能垒;带隙较小可能改善光吸收或输运,也可能带来复合、稳定性或选择性问题;Bader 电荷变化提示分区电子数改变,价态和反应活性仍由配位、轨道和能量证据共同限定。
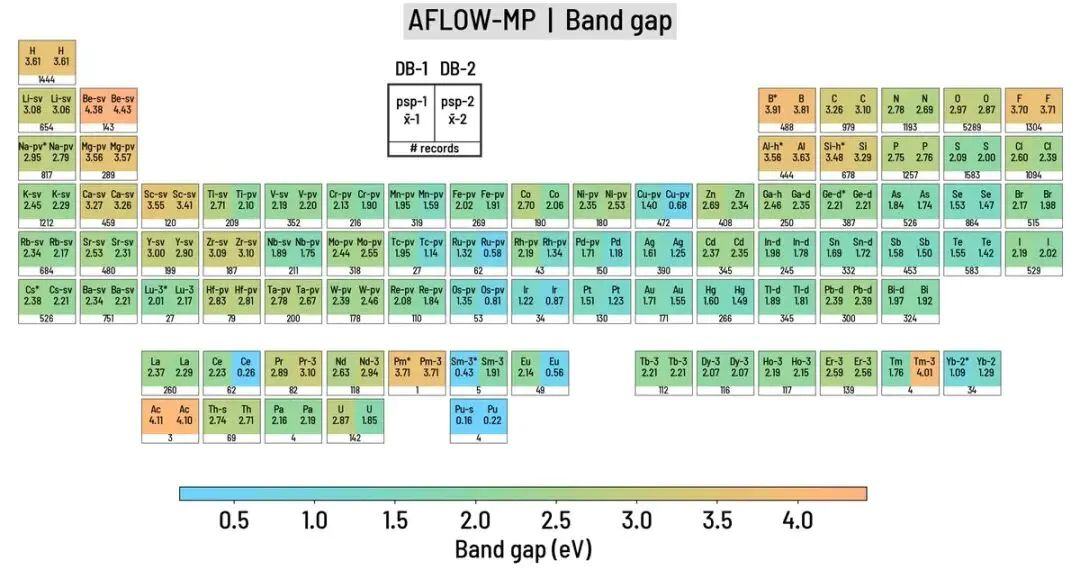
图7. AFLOW-MP 对比中,按元素统计的化合物带隙中位值和赝势标识。DOI:10.48550/arXiv.2007.01988
带隙的元素分布图给出一个典型例子:同样是 DFT 数据库,同一元素相关化合物在不同数据库中的带隙统计存在差别。材料筛选时若只保留“带隙大/小”这样的标签,泛函、赝势、磁性和结构样本的影响会被抹平。结论与具体物理对象绑定后,“第一性原理计算表明”才对应可复查的能量、电子态或结构变化。
在材料计算里,第一性原理指向从原子核和电子出发构造材料问题的计算方式:输入是元素、结构和约束条件,求解对象是电子结构与能量面,输出是能量、力、密度和图谱。材料机制中的假设需要转化为可检查的模型量,每个判断都对应具体计算条件、比较对象和参考态。