说明:本文华算科技主要介绍吸附能的定义式、参考态选择、结构模型来源、自由能修正和结果使用边界。
吸附能的基本定义
吸附能 Eads 本质上是一个总能差,它比较的是吸附物靠近表面并形成稳定吸附态前后的能量变化。常见写法为 Eads = Eslab+ad − Eslab − Ead。Eslab+ad、Eslab 和 Ead 三项必须成套定义,分别对应吸附态总能、清洁基底总能和孤立吸附物总能。符号方向要跟原文定义一致:按这个写法,负值通常表示吸附态相对分离态更低能;若文献把结合能写成相反号,正值反而代表更强相互作用。
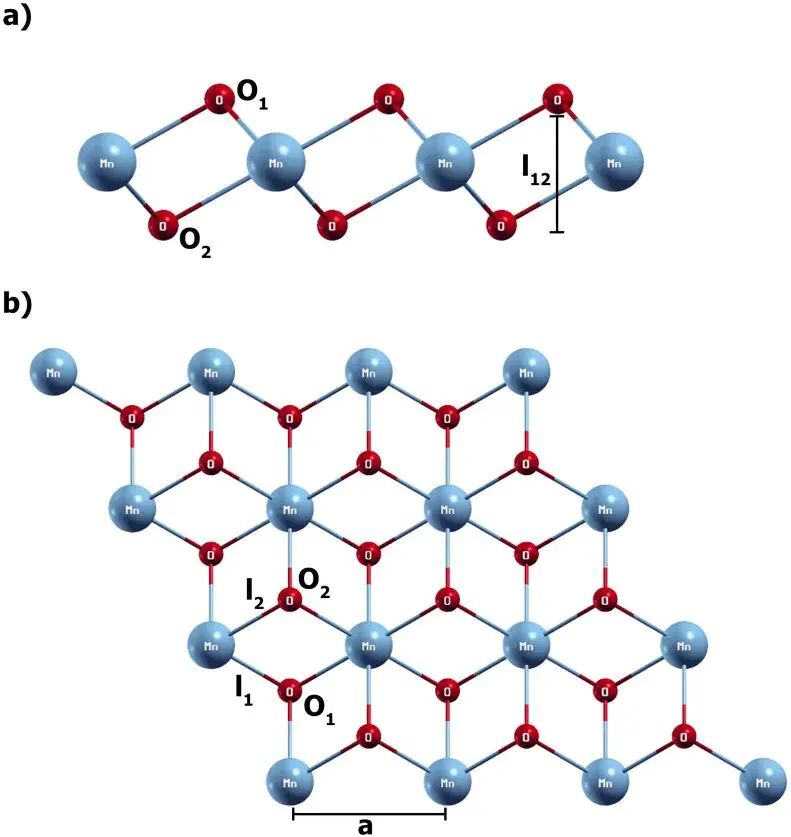
图1. 1T-MnO2 单层的俯视与侧视结构,用于定义吸附前基底总能 Eslab。DOI:10.1371/journal.pone.0335153
公式看起来很短,真正容易出错的是三个能量项是否来自同一计算层级、同一元素赝势、同一泛函和可比的参考态。清洁表面要先优化到目标模型状态,孤立分子通常放在足够大的真空盒中,开壳层分子还涉及自旋态。O2、NO、自由基、过渡金属原子这类参考态对自旋和泛函更敏感,Ead 的误差会原样传入 Eads。
表面吸附并不是把一个分子随便贴到晶面上。吸附能只属于某个明确构型:哪一个晶面、哪一种终止、哪个吸附位、覆盖度多少、吸附物是分子态还是解离态,这些条件改变后,Eslab+ad 也跟着改变。单原子吸附、分子吸附、共吸附和解离吸附的参考式可以写得相似,但参考对象不同,不能把它们的数值直接排在一张表里当同一类指标。
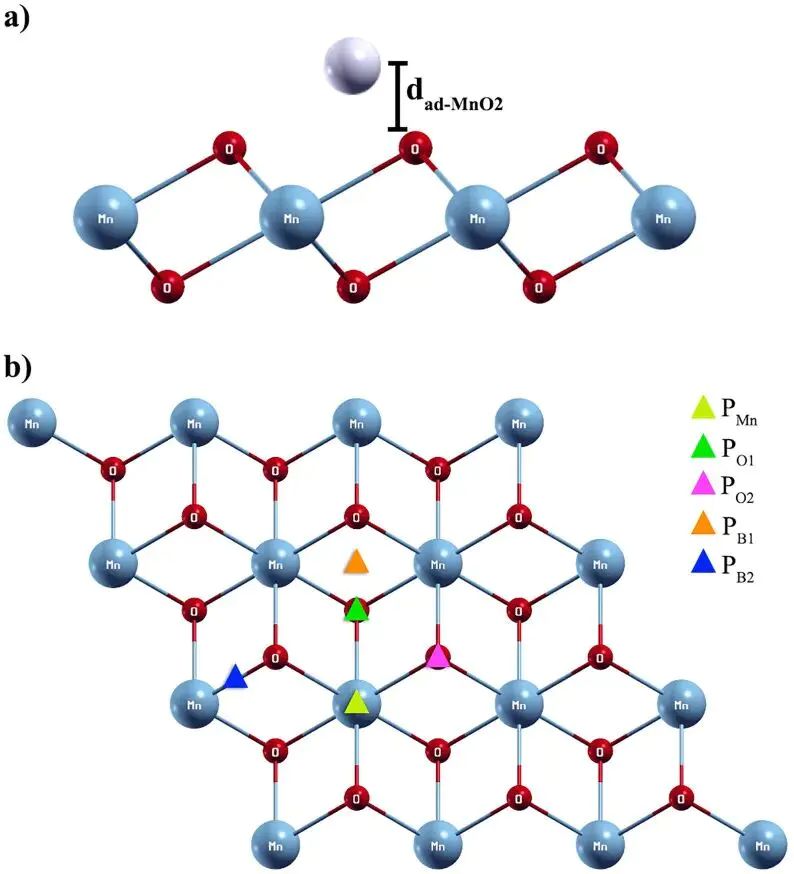
图2. Pb/Cd 在 1T-MnO2 表面的非等价吸附位点与吸附距离定义。DOI:10.1371/journal.pone.0335153
若表面上有 n 个相同吸附物,常见处理是计算总吸附能后再除以 n,得到每个吸附物的平均吸附能。对氢吸附常写 Eads,H = [Eslab+nH − Eslab − n/2 EH2]/n。这个式子把 H2 作为氢化学势参考,适合 HER 相关比较;若讨论原子氢束流、等离子体或缺陷捕氢,参考态就可能改变。参考态一变,吸附能的零点也跟着移动,数值大小要回到具体反应环境中解释。
吸附能计算需把控哪些步骤?
计算吸附能时,第一步是给清洁表面建立可以承载吸附过程的模型。晶面指数、层数、真空层厚度、底层是否固定、表面终止、超胞大小都会影响表面局域电子结构。Eslab 不是抽象常数,它来自某个已经弛豫好的基底模型;模型太薄、真空层太小或偶极修正缺失时,后续吸附能会混进人为相互作用。
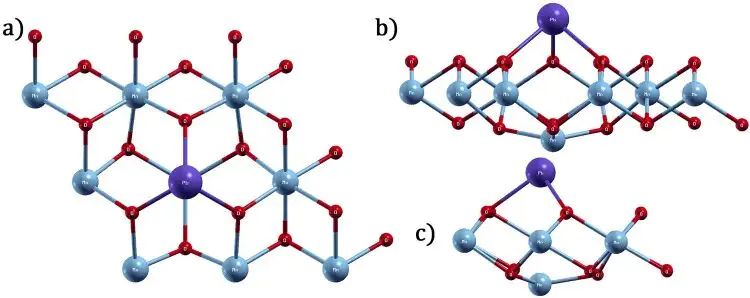
图3. Pb 吸附后 1T-MnO2 单层的优化结构,呈现吸附态总能 Eslab+ad 对应的几何对象。DOI:10.1371/journal.pone.0335153
第二步是枚举有物理意义的吸附构型。顶位、桥位、空位、缺陷附近、边缘位、界面位都可能给出局部极小值。对金属氧化物、二维材料和单原子催化位点来说,吸附物轻微偏转就可能改变配位数和轨道杂化方式。只优化一个初始构型往往不能代表全局较低能吸附态,特别是分子可以旋转、解离或跨位点成键时,吸附能比较必须带着构型筛选结果一起出现。
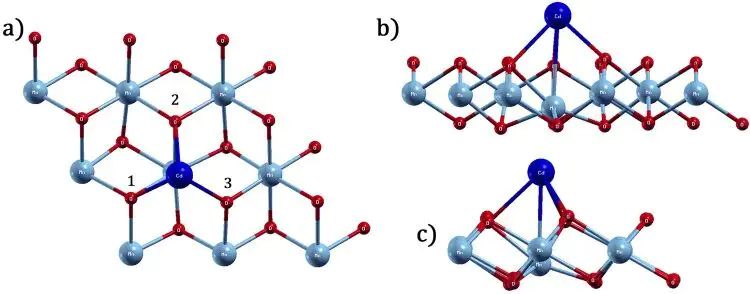
图4. Cd 吸附后 1T-MnO2 单层的优化结构,不同吸附物会给出不同键长和局部弛豫。DOI:10.1371/journal.pone.0335153
第三步是把吸附态结构充分优化,得到 Eslab+ad。吸附后表面原子可能向外拔起,也可能发生局部重构;吸附物内部键长、键角和电荷分布也会变化。若只固定吸附物位置做单点能,算到的是某个外加几何约束下的能量,并不等价于稳定吸附态。吸附能计算的对象是弛豫后的能量差,几何弛豫不足会把力没有释放完的结构误差写进能量。
最后一步是把能量项按相同收敛标准代入公式。k 点、截断能、电子收敛、弛豫阈值、色散修正、U 值、赝势版本都要保持可比。对于带电吸附、极性表面或外电场体系,还要检查电荷补偿、偶极修正和电势零点。所谓“怎么算”,实际是在给同一物理过程建立一组可比较的能量账本,不是只把三个输出文件里的能量相减。
差分电荷密度常与吸附能放在同一讨论单元中,但它回答的是电子重排位置和方向。电荷在吸附物与基底之间聚集,通常对应配位成键、轨道杂化或界面电场变化;电荷耗尽区则提示原有电子云被拉开。差分电荷不能直接换算成吸附能大小,它更适合解释为什么某个构型产生了较强或较弱的相互作用。
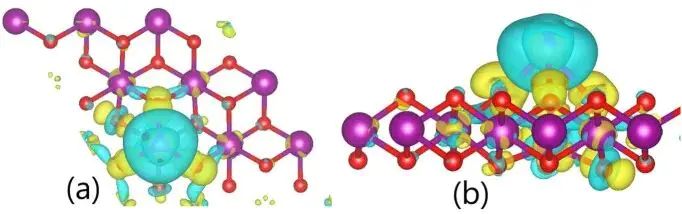
图5. Cd 吸附在 1T-MnO2 后的差分电荷密度等值面,用于定位界面电子重排。DOI:10.1371/journal.pone.0335153
吸附能数值为何不具备绝对性?
同一个 Eads 公式,在不同研究中出现差别,最常见的原因是参考态和计算方法不同。以气相分子为参考时,Ead 包含分子本身的键能、磁性和零点振动;以电化学氢电极、溶液物种或金属体相为参考时,能量零点已经改变。吸附能不是绝对标尺,它只在参考态一致的比较组内有清楚含义。
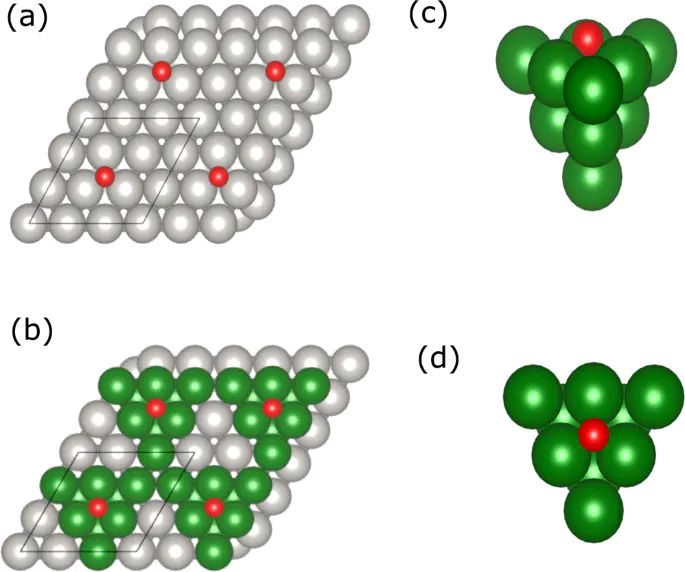
图6. 从 O/Pt(111) 周期吸附结构中抽取簇模型的示意图,呈现周期模型与簇校正之间的对应。DOI:10.1038/s41467-022-34507-y
泛函选择是另一个主要来源。PBE、RPBE、SCAN、杂化泛函以及色散修正对金属表面、弱吸附分子和强化学吸附键的描述并不相同。某个泛函下更负的吸附能,不一定在更高层级方法下仍然保持同样排序。文献中用簇模型校正周期吸附能,就是为了把局部化学键部分拿到更高精度层级评估,再把差值反馈给周期模型。
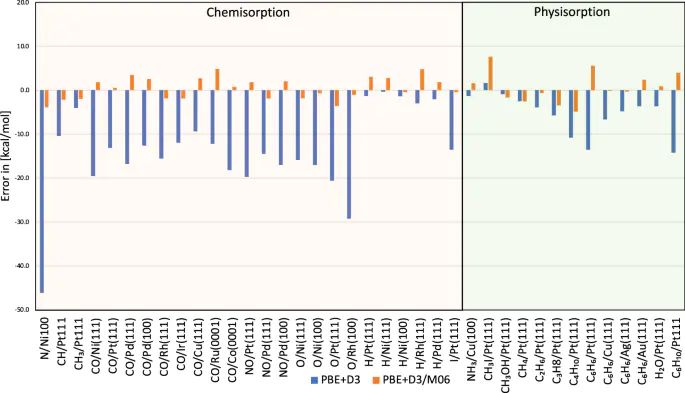
图7. 化学吸附与物理吸附体系中,PBE+D3 吸附能误差及簇校正后的误差对比。DOI:10.1038/s41467-022-34507-y
当覆盖度升高、相邻吸附物距离变短时,Eads 会把静电排斥、氢键、偶极耦合或表面应变一起计入能量差。低覆盖度下,吸附物之间距离较远,Eads 更接近单个吸附事件;覆盖度升高后,数值会带上相邻吸附物之间的相互作用。若把高覆盖度结果拿去代表稀薄吸附,数值里已经包含吸附物之间的相互作用,这时它不再只是单个分子和单个位点的成键强度。
把电子总能用于真实反应环境时,温度、压力和溶剂模型会把 Eads 推向自由能问题。气相催化常引入零点能、热容和熵修正,电催化还会牵涉 pH、电极电势、双电层、显式水和恒电势条件。电子能吸附强弱、Gibbs 自由能、反应中间体稳定性属于不同层级,不能把没有修正的 Eads 直接当作工作电位下的反应自由能。
金属、氧化物、硫化物和二维材料对上述因素的敏感程度也不同。金属表面常见 d 带中心、配位数和应变调控;氧化物表面还涉及氧空位、价态变化和 Hubbard U;二维材料则要注意层间相互作用和表面偶极。吸附能差异背后通常是局域电子结构、配位环境和参考体系共同改变,单独盯住一个数字很容易把方法误差和真实材料差异混在一起。
吸附能结果怎样进入材料判断?
吸附能最常见的用途,是比较一组材料或位点对同一吸附物的稳定能力。合理比较的前提是计算对象一致:同一种中间体、同一种参考态、相近覆盖度、相近收敛标准和明确构型筛选。吸附能更负只能说明该参考框架下吸附态更稳定,它不能单独推出催化活性更高、选择性更好或循环稳定更强。
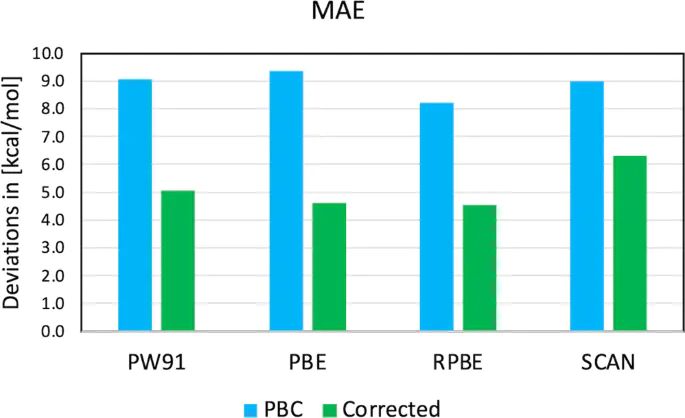
图8. PW91、PBE、RPBE 和 SCAN 周期吸附能在校正前后的平均绝对误差对比。DOI:10.1038/s41467-022-34507-y
在催化反应里,中间体既要能生成,也要能离开表面继续下一步反应。过弱吸附可能导致反应物难以活化,过强吸附则可能占据活性位或提高产物脱附代价。更合理的判断方式,是把吸附能放到整个反应能量图中,看相邻步骤的能量差、能垒和产物脱附是否匹配。火山关系的核心也不是追求越强越好,而是寻找中间体稳定性与后续转化之间的平衡区间。
吸附能还常用于判断构型可信度。若某个位点给出明显更低的 Eads,并且对应结构具有合理键长、差分电荷呈现界面电子重排、PDOS 或 COHP 指向轨道杂化和反键态占据的改变,那么该构型更有可能成为主要吸附态。反过来,若能量差只有几 meV,却伴随不同自旋态、不同收敛精度或不同溶剂模型,排序就不宜写成确定判断。
对于电催化和电池界面,吸附能尤其要注意工作环境。真空表面上算出的 OH*、O*、CO* 或 Li* 吸附能,能帮助识别本征成键趋势;进入电解液、电极电势和离子浓度环境后,界面电场、双电层屏蔽和溶剂化会重塑中间体稳定性。Eads 可以作为机制坐标,但完整判断常要联合自由能、Bader 电荷、态密度、反应能垒和表面稳定性。
实际做材料筛选时,吸附能适合回答“这个位点是否容易抓住该中间体”“不同取代或缺陷是否改变局域成键”“哪类表面可能处在过强或过弱吸附区间”。它不适合独自回答“材料一定更好”这类问题。若比较不同晶面,应标明表面能、重构和暴露稳定状态是否已经分开处理;若比较不同中间体,参考分子和自由能修正也要标明。把吸附能当作能量账本中的一项,它才会成为判断材料机制的可靠坐标。