本华算科技了的理论基础、核心计算方法(如、CASSCF和耦合簇理论)及其在光化学反应、OLED材料设计和生物成像等领域的关键应用,如何通过计算模拟研究激发态性质,从而高效设计发光材料、揭示光化学机理并优化生物探针性能。
什么是分子激发态
理解分子激发态是现代光化学、材料科学和生命科学的核心,其理论根植于量子力学的基本框架。
量子力学基础
薛定谔方程该方程的解——波函数,包含了体系的所有信息。对于激发态,需求解与时间相关的薛定谔方程或采用变分原理对波函数进行优化,以得到激发态的能量与性质。
量子力学的不确定性原理在理解激发态的动力学行为中也扮演着关键角色。
激发态的基本特性
这些弛豫途径包括:为描述激发态的产生和性质,科学家发展出多种理论模型。例如,分子激子理论,其中包括Frenkel激子和Davydov分裂等概念,用于解释分子聚集体中的激发态行为。这些理论为理解光谱特征和能量传递奠定了基础。
激发态计算的核心方法
时间依赖密度泛函理论 (TD-DFT)
其最大优点是计算效率高,适用于中等乃至较大的分子体系,能够较好地预测激发能、几何结构和振子强度然而,它也存在明显局限:指出TD-DFT难以准确描述多参考态特征明显的体系(如双激发态)、长程电荷转移激发态以及Rydberg态,存在一定的系统性误差。它常作为快速筛选工具或用于对精度要求不极高的场景。
完全活性空间自洽场方法 (CASSCF)及其扩展
它通过定义活性空间,能对复杂电子结构(如光化学反应中的锥形交叉点CI)进行高精度描述。
耦合簇系列方法是高精度计算的重要选择。除上述方法外,还有配置相互作用单重态(CIS),计算快速但精度较低,可用于大分子的初步研究;多参考组态相互作用(MRCI)提供高精度但成本巨大;
近年来,机器学习(ML)技术也被用于快速预测激发态性质和开发新泛函多体格林函数理论(MBGFT)则被应用于扩展体系的光学性质计算。
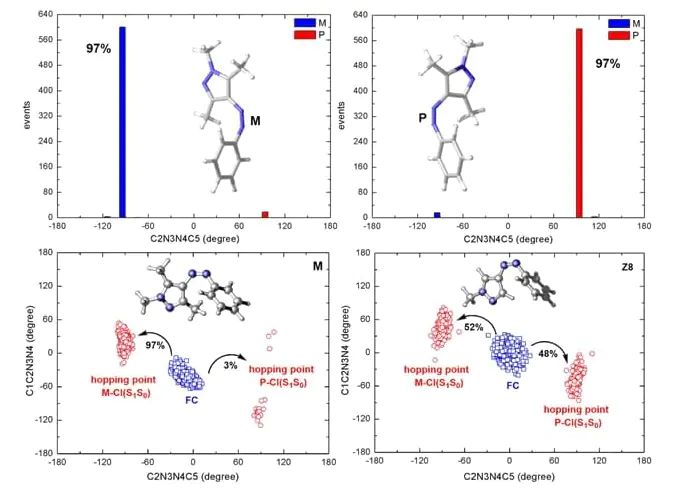
计算模拟是揭示光化学反应机理的“计算机显微镜”。量子化学例如,arylchlorodiazirines类分子光解反应的理论研究₁态激发能(在有机发光二极管(OLED)材料设计中,激发态计算发挥着至关重要的作用。例如单重态-三重态能隙(ΔE-ST)、辐射/非辐射衰减速率、自旋-轨道耦合(SOC)矩阵元等,从而预测材料的发光效率、发光颜色和机制(如荧光、磷光、热激活延迟荧光TADF)。这为设计高效率、长寿命的TADF材料提供了直接的理论指导。
生命科学与生物成像
计算模拟(如极化嵌入(PE)模型与代数图解构造(ADC)方法结合)可用于研究生物大分子中的电荷转移激发态,理解其光谱特征并指导新型荧光探针的开发。例如,对绿色荧光蛋白(GFP)发色团激发态的精确计算,帮助揭示了其发光机理并指导了突变体设计。
结论
从高精度的CASSCF/CASPT2和耦合簇方法到高效实用的TD-DFT,计算方法的不断进步使得我们能够以前所未有的细节洞察光与物质相互作用的微观世界。