:计算是研究的核心工具,可解析反应机制、建立活性描述符、指导设计。其关键内容包括、、等,通过顶刊案例验证了对、等反应的机制阐释与性能预测能力。
DFT计算在电催化研究中,DFT,其核心作用体现在三个相互关联的维度。
DFT。通过计DFT计算氢电极(CHE)模型的引入,进一步将电极电位(U)与反应自由能关联,使DFT能直接模拟不同电势下的反应行为,实现过电位的定量预测。

其次DFT,实现在电子结构描述符方面,带中心电荷分布分析则量化了单原子催化剂中金属–载体的电荷转移量,这种电荷分布与ORR活性呈线性相关,为SACs的活性优化提供了明确指标。
DOI:10.1360/nso/20220059
,为催化剂的理性设计提供了从活性位点识别到高通量筛选的全流程指导性位点识别中,可定位催化剂的真实活性中心,例如MoS₂的HER活性并非来自基面,而是边缘的硫空位,明确了缺陷工程的优化方向。
高通量筛选此外,DFT能量化异质结的界面效应,BiFeO₃/BiVO₄界面的电荷重分布计算,内置电场强度达0.2 V/Å,加速光生电子–空穴分离,解释了其优异的光催化水分解活性。

DFT计算的关键内容
在电催化中的应用依赖于一系列核心计算模块的协同,这些模块从不同角度捕捉催化反应的关键特征,并通过与实验的协同验证,构建起理论与实践的桥梁。核心计算模块涵盖五个相互关联的内容,每个模块都有明确的物理意义和顶刊应用案例。
(ΔG_*ads)中间体与催化剂表面结合强度的核心参数,其值直接决定反应能垒与活性——ΔG_*ads过强或过弱都会导致动力学迟滞。
DOI:10.32657/10356/73265
TS通过NEB电子结构分析通过计算带中心、态密度(DOS)及电荷布居示催化剂活性的电子起源。例如,在单原子催化剂()中,DFT计算的d带中心偏移与O吸附能的线性关系,直接关联电子态与催化活性,Nature Materials的工作据此解释了Fe-N₄位点的高ORR活性。

溶剂化效应校正模拟固液界面真实环境的必要步骤,通过隐式溶剂模型或显式水分子层,修正溶剂极性对吸附能的影响,如的研究显示,显式水分子可使Pt表面OH的吸附能降低0.15 eV,更贴近实验条件。
Pourbaix通过UDOI:10.1039/d2cp04785c
DFT与实验的协同验证模式进一步提升了理论计算的可靠性,形成“计算指导实验,实验验证计算”的闭环。
方面NiMoN/NiFe LDH构效关联XPS性能预测,预测Cu-M双金属催化剂中,M的掺杂可降低CO₂→C₂H₄的决速步能垒,实验中该催化剂的C₂H₄法拉第效率达80%,印证了DFT的预测能力。

DFT顶刊应用
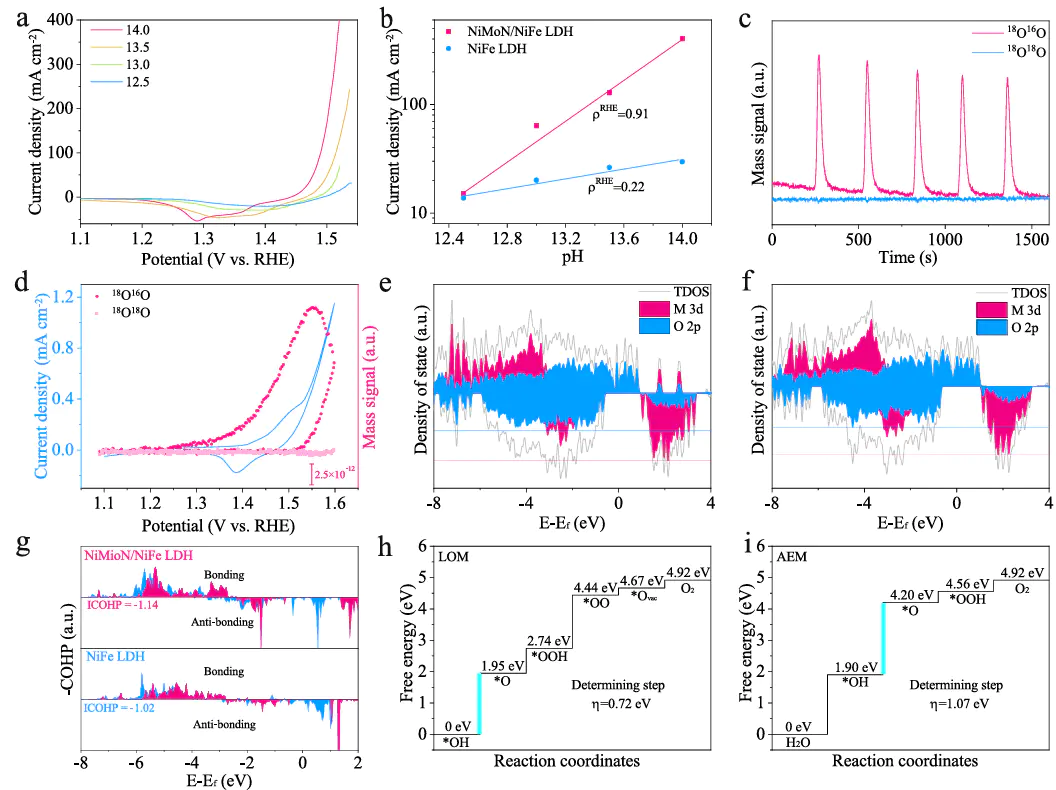
Natureitride/hydroxide for oxygen evolution at large current density”中,计算作为核心研究手段,从电子结构、反应路径到动力学验证,全方位揭示了NiMoN/NiFe LDH异质结在高电流密度下的OER活性起源,展现了顶尖研究中DFT的深度应用DOI:10.1038/s41467-023-37091-x
焦,DFTd带中心的上移增强了Ni位点与O中间体的轨道杂化,使O吸附能从-3.2 eV优化至-3.5 eV,加速了O-O耦合步骤,为高活性提供了电子结构基础。
是该研究DFT在AEM路径中,*O→*OOH步骤的自由能变为1.58 eV,成为决速步;而在LOM路径中,晶格氧参与反应,*O_lattic→*O₂步骤的ΔG为0.87 eV,能垒显著降低。
电子密度示,LOM路径中NiMoN的电子转移使O_lattic的电子云密度降低,弱化了M-O键,促进O-O键形成。动力学验证通过微动力学模型实现,基于DFT计算的能垒数据,模型预测LOM路径的速率常数是AEM路径的10³倍,明确LOM为高电流密度下的主导路径。
突破了传统静态计算的局限,通过构建动态表面模型,解决了理论与实验的偏差问题传统静态模型假设表面清洁,而实际高电流密度下,的覆盖率达80%,DFT计这种动态模型的引入,使DFT计算更贴近工业级电流密度的真实反应环境,为非贵金属催化剂在实际工况下的性能预测提供了新范式。
该研究通过指导的界面电子态调控策略,实现了高电流密度下的低过电位,展示了DFT从机制解析到设计策略的全链条价值成为“理论–计算–实验”融合的典范。
电催化DFT计算的发展
DFT动态过程模拟是应对催化剂表面重构的关键进展DFT动力学(MD)与DFT的耦合(AIMD)可实时模拟表面重构,如FeOOH在OER过程中的动态空位形成——AIMD模拟显示,高电位下Fe-O键断裂形成氧空位的能垒从静态计算的1.8 eV降至1.2 eV,空位处的O吸附能优化0.3 eV,解释了实验中观察到的活性提升。
DOI:10.1002/smsc.202100011
DFT,神DFT在高熵合金筛选中,可实现亿级构型的快速采样,结合DFT验证,从10万种可能组成中筛选出CoCrNiMnAl的OER过电位低于IrO₂。
DOI:10.1039/d2sc06403k
DFT,传统隐式溶剂模型(如)难以捕捉水分子的具体作用,而显性水分子层与针对高离子强度电解液,DFT结合泊松–玻尔兹曼方程(PBE)处理离子分布,如Li⁺在NiFe LDH表面的吸附使OOH的ΔG降低0.2 eV,解释了Li⁺添加剂的活性提升作用。

多尺度建模实现了从原子尺度到宏观反应器的全链条模拟将计算的电子结构数据输入介观动力学蒙特卡洛(kMC)模型,可模拟表面覆盖度随时间的演化,如Pt纳米颗粒上ORR的kMC模拟显示,*OH覆盖率达50%时活性最佳。
DOI:10.1021/acs.accounts.2c00058
在电催化研究中DFT,其在基础研究层面,通过原子级的电子结构与能量计算,揭示了反应中间体吸附、电荷转移、过渡态形成等微观过程,建立了“电子态–吸附能–反应能垒”的关联,使电催化机制从定性推测走向定量描述。
高通量筛选机器学习催化剂。
的独特价值还体现在其对实验的“反向指导”作用仅能解释实验现象,更能通过电子层面的设计提出颠覆性策略。当前,随着动态模拟、溶剂化模型革新及多尺度耦合的发展,正逐步克服静态计算、简化模型的局限,更真实地模拟实际反应环境,其预测精度与实验的吻合度持续提升。